3 The STI1-domain is a flexible alpha-helical fold with a hydrophobic groove
Adapted from Fry MY*, Saladi SM*, Clemons WM. 2021a. The STI1-domain is a flexible alpha-helical fold with a hydrophobic groove. Protein Sci 30:882–898. doi:10.1002/pro.4049
3.1 Abstract
STI1-domains are present in a variety of co-chaperone proteins and are required for the transfer of hydrophobic clients in various cellular processes. The domains were first identified in the yeast Sti1 protein where they were referred to as DP1 and DP2. Based on hidden Markov model searches, this domain had previously been found in other proteins including the mammalian co-chaperone SGTA, the DNA damage response protein Rad23, and the chloroplast import protein Tic40. Here, we refine the domain definition and carry out structure-based sequence alignment of STI1-domains showing conservation of five amphipathic helices. Upon examinations of these identified domains, we identify a preceding helix 0 and unifying sequence properties, determine new molecular models, and recognize that STI1-domains nearly always occur in pairs. The similarity at the sequence, structure, and molecular levels likely supports a unified functional role.
Keywords: Protein targeting, co-chaperones, Ubiquilins, Sti1, Hop, SGTA, HIP, UBL-UBA
3.2 Introduction
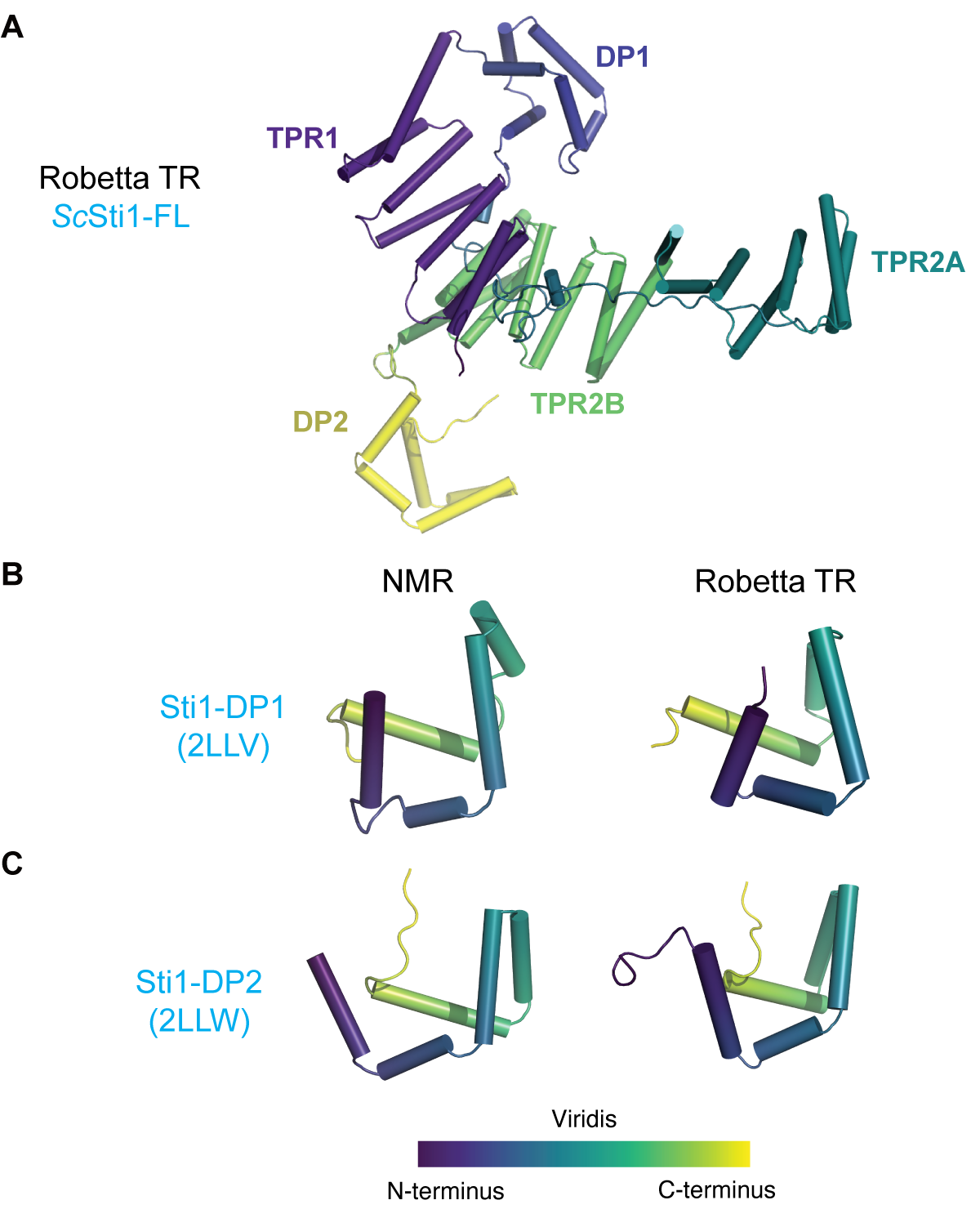
Hydrophobic stretches that are exposed during protein biosynthesis can aggregate which poses a risk to cellular homeostasis. To avoid this, cells have evolved proteins that bind to and protect hydrophobic segments. Several protein domains exist to assist these proteins and are found in protein families that occur broadly in eukaryotes, here we focus on the STI1-domain. Named for the yeast protein Sti1 (STress Inducible 1) where they were first identified, STI1-domains are referred to as heat-shock chaperonin-binding domains in databases (Letunic and Bork, 2018). Solved structures from Sti1 revealed an alpha-helical domain with five amphipathic helices that present a hydrophobic groove, an likely binding site for hydrophobic segments of a client (Li et al., 2013; Schmid et al., 2012) (Figure 3.1A,B). In addition to Sti1 homologs, a number of protein families were bioinformatically identified to contain this domain including the co-chaperones HIP (HSP interacting protein) and SGTA (Small Glutamine-rich TPR-containing protein A), the DNA damage response protein Rad23 (RADiation sensitive 23), yeast UBL-UBA family member Dsk2 (Dominant Suppressor of Kar1 2), human KPC2 (Kip1 ubiquitylation-Promoting Complex 2), human ubiquilins (UBQLNs) 1-4, and the plant chloroplast import protein Tic40 (Translocon at the of the Inner envelope membrane of Chloroplasts 40) (Howe et al., 2020; Zientara-Rytter and Subramani, 2019). The identified STI1-domain containing proteins can be broadly classified into two categories: either co-chaperones (homologs of Sti1, HIP, and SGTA and the unique plant Tic40) or adaptors of the ubiquitin proteasome system (AUPS) (homologs of the mammalian Rad23, UBQLNs, KPC2, and yeast Dsk2).
STI1-domain containing proteins have been identified only in eukaryotes. HOP and Rad23 are found throughout eukaryotes including in some protists. SGTA homologs are similarly prevalent, although not readily identifiable in Viridiplantae (Howe et al., 2020). UBQLNs, the closest mammalian relatives to yeast Dsk2, can be found across multicellular eukaryotes (Howe et al., 2020; Zientara-Rytter and Subramani, 2019). Tic40 is restricted to Archaeplastida (algae and land plants). It is likely that more distant homologs for each of these STI1-domain containing proteins exist in taxa that currently seem excluded (Weisman et al., 2020).
We consider the co-chaperones first. Broadly, co-chaperones are binding partners for Hsp90 or Hsp70 that enhance the function of these chaperones with a subset directly involved in binding to clients (Caplan, 2003). A first example is the mammalian Sti1-homolog HOP (Hsp70/Hsp90 organizing protein) that coordinates the essential transfer of clients between the chaperones Hsp70 and Hsp90. The abundant chaperone Hsp90 and its homologs are involved in multiple cellular pathways and many clients are first loaded from homologs of the chaperone Hsp70 (Prodromou, 2016). The domain organization of HOP homologs includes two STI1-domains, originally named DP1 and DP2 due to a repeated DP motif(Chen and Smith, 1998; Prapapanich et al., 1998), that are preceded by Hsp70/90-binding tetratricopeptide-repeat (TPR) domains (Kajander et al., 2009; Nelson et al., 2003; Onuoha et al., 2008; Schmid et al., 2012) (Figure 3.1A). In yeast, in vivo deletion of the second STI1-domain in Sti1 (DP2) is detrimental, impairing native activity of the glucocorticoid receptor (Schmid et al., 2012). In vitro, removal of DP2 results in the loss of the transfer of the progesterone receptor to Hsp90 (Nelson et al., 2003). These results implicate DP2 in client interaction. Besides simply bridging client transfer between the two HSPs, HOP has also been implicated in prion-protein binding (da Fonseca et al., 2021; Martins et al., 1997; Zanata et al., 2002). Like HOP, HIP also aids in the transfer of client from Hsp70 to Hsp90 through direct interaction with Hsp70 (Lässle et al., 1997; Prapapanich et al., 1996; Reidy et al., 2018).
For the mammalian SGTA and its homologs, including yeast Sgt2, the STI1-domain directly binds to clients (Lin et al., 2021). SGTA homologs play a number of roles, with the best characterized involving the targeting of tail-anchored (TA) proteins to the ER membrane as a member of the Guided Entry of TA protein (GET) pathway (Chartron et al., 2012, 2011; Rao et al., 2016; Simon et al., 2013; Wang et al., 2010). SGTA has been suggested to play a role in the degradation of mislocalized membrane proteins in conjunction with the protein Bag6 (Hessa et al., 2011; Leznicki et al., 2015; Mock et al., 2015; Rodrigo-Brenni et al., 2014; Wunderley et al., 2014; Xu et al., 2012). Additionally, SGTA is involved with disease, including polyomavirus infection (Dupzyk et al., 2017), neurodegenerative disease (Kiktev et al., 2012; Long et al., 2012), hormone-regulated carcinogenesis (Buchanan et al., 2007; Trotta et al., 2013), and myogenesis (Wang et al., 2003), although the underlying molecular mechanisms are still unclear.
The final member of the co-chaperone family is the chloroplast protein Tic40. In Arabidopsis thaliana, Tic40 is found in the inner membrane of the chloroplast and has been suggested to be a co-chaperone for the stroma chaperone complex for protein transport across the inner membrane (Bédard et al., 2007; Chou et al., 2003). Deleting Tic40 leads to a decrease in the import of precursors into the chloroplast (Kovacheva et al., 2005). Where studied, STI1-domains in each co-chaperone interact with clients (Chang et al., 1997; Chartron et al., 2011; Fan et al., 2002; K. Ko et al., 2004; Li et al., 2013; Lin et al., 2021; Mock et al., 2015; Nelson et al., 2003; Schmid et al., 2012; Wang et al., 2010), thus due to the role of the STI1 motif in other co-chaperones, the STI1-domain in Tic40 may also interact with clients being transported across the outer chloroplast membrane and into the stroma (Bédard et al., 2007; Chou et al., 2006, 2003; Kovacheva et al., 2005).
The second group of STI1-domain containing proteins, the AUPS, primarily deliver clients to the proteasome. They contain an N-terminal ubiquitin-like (UBL) domain and a C-terminal ubiquitin-associated domain (UBA). One of the earliest identified UBL-containing protein in yeast was Rad23; this protein shuttles some proteins to the proteasome and also protects some clients from degradation by preventing ubiquitin elongation (Elsasser et al., 2002; Fishbain et al., 2011; Saeki et al., 2002; Wade and Auble, 2010; Watkins et al., 1993). Rad23 has also been implicated in nucleotide excision repair as a complex with Rad4 that recognizes DNA damage (Dantuma et al., 2009; Zientara-Rytter and Subramani, 2019). Likewise the fungal Dsk2 acts as an adaptor to target ubiquitin-labeled proteins to the proteasome for degradation (Lowe et al., 2006). The UBA domain of Dsk2 recognizes the poly-ubiquitin tail on proteins and the UBL domain interacts with the proteasome regulatory subunit, Rpn1. Another UBL-UBA containing adapter protein is KPC2 (Hara et al., 2005), a subunit of the KPC E3 ligase complex where it acts as an adapter for p27 ubiquitination in the G1 phase of the cell cycle (Kotoshiba et al., 2005).
The closest mammalian homologs to Dsk2 are the four ubiquilins, UBQLN-1 to 4 (Zientara-Rytter and Subramani, 2019). While UBQLN-1 is universally expressed and UBQLN-2 & -4 are expressed in most tissues, UBQLN-3 is expressed only in the testes (Conklin et al., 2000; Yuan et al., 2015). The best characterized of these, UBQLN-1, functions similar to Dsk2 and Rad23 by delivering poly-ubiquitinated proteins to the 26S proteasome. Other demonstrated roles for UBQLN-1 are an association with aggregates for delivery to the lysosome for degradation and in the ER-associated degradation (ERAD) pathway (El Ayadi et al., 2013; H. S. Ko et al., 2004; Lim et al., 2009; Zhang et al., 2008). In UBQLN-1, STI1-domains have been shown to bind to TMDs of mitochondrial membrane proteins and target them to the proteasome for degradation (Itakura et al., 2016). The direct involvement of UBQLNs in client degradation suggests a broader role than simply being shuttling factors (Itakura et al., 2016).
Here, we inspect these identified STI1-domains and the proteins they reside in to clarify the criteria for this domain. Upon examination, there are clear similarities in the structural features of most STI1-domains, while some currently defined STI1-domains are likely misannotated. Based on structure-based sequence alignments and similarity in predicted secondary structure, we develop a new definition that has allowed the identification of other STI1-domains and clarification of previously misannotated domains. We employ structural prediction methods to model uncharacterized STI1-domains revealing a consistent alpha-helical hand architecture. When considering proteins that contain STI1-domains, we find similar functional roles and domain architecture. In total, this work provides a comprehensive definition of the STI1-domain.
3.3 Results
3.3.1 Amalgamating and examining predicted STI1-domains
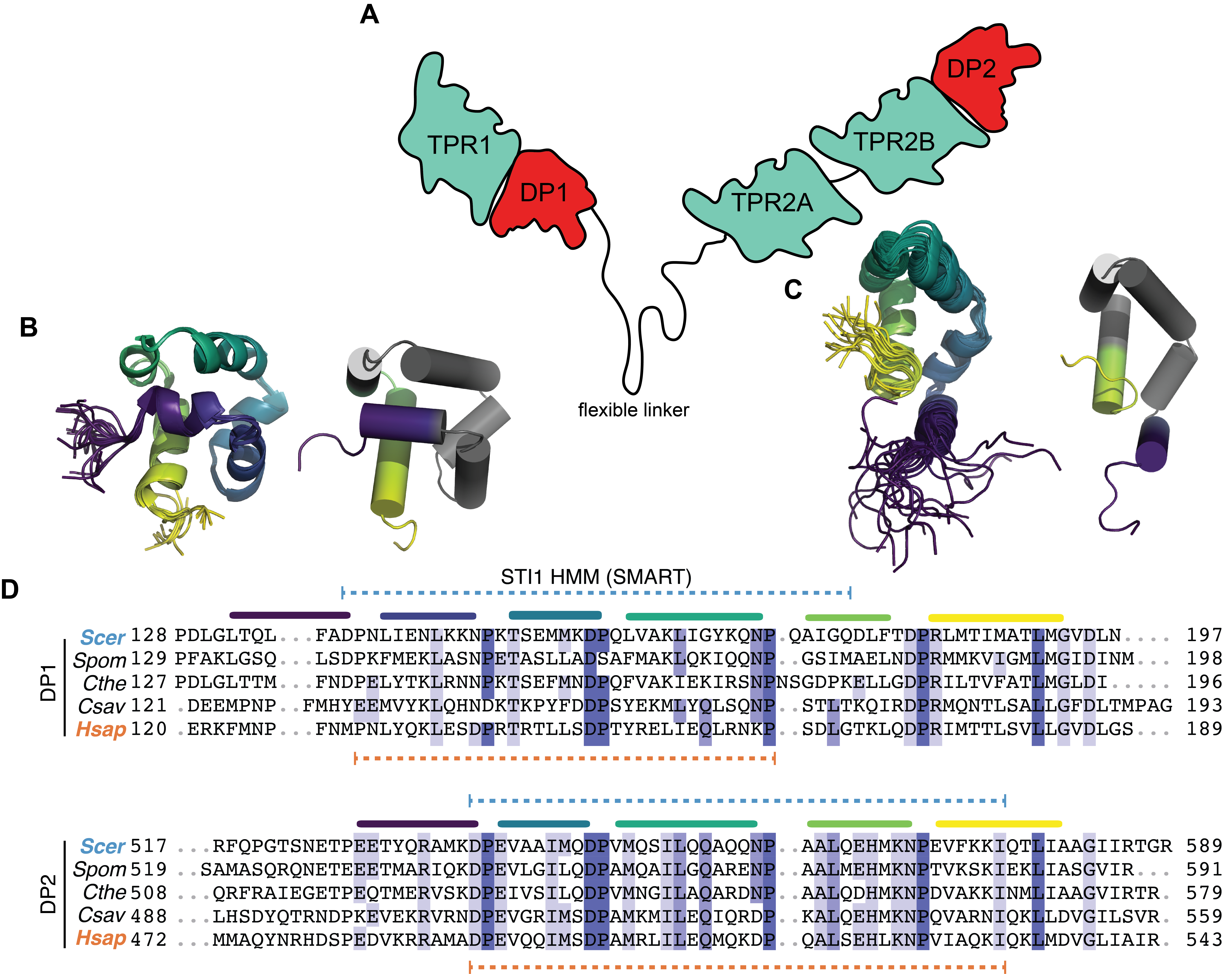
A search through protein databases shows a variety of entries with a name or protein domain annotation that includes “Sti1”. Close homologs of HOP have “Sti1” or “Sti1-like” in their entry name. These are typically bidirectional BLAST best-hits, i.e., for a new sequence, the top scoring hit in a reference database identifies the original sequence when searched against the set of new sequences (Ward and Moreno-Hagelsieb, 2014). Other entries also have “Sti1” domain annotation. This annotation originates from a hit to the STI1 Hidden Markov Model (HMM) created by the Simple Modular Architecture Research Tool (SMART) database (since 2001) (Letunic and Bork, 2018) and more recently from an HMM in the Pfam v32 database (El-Gebali et al., 2019) (Figure 3.1D). The STI1-domain is named as such due to the prevalence of Sti1 homologs in the seed sequences of the SMART and Pfam HMMs. HMM-based methods reliably identify homologs of lower sequence similarity and are much more sensitive than sequence-to-sequence searches such as BLAST. HMM-based searching achieves higher sensitivity by using an alignment of query sequences (a “seed”) to search a target database. Typically, the set of protein sequence regions that comprise seeds are curated by hand, but the creators of SMART developed an automated method to compile seed protein regions for the categorization of protein domains distinct from those identified by human curators, as in the case of Pfam.
The SMART HMM for the STI1 family provides a useful initial annotation, yet when comparing the HMM to known structures and sequence alignments of STI1-domains, several issues come to light. The first being a partial hit for each DP domain in Sti1 where only four of the five helices are recovered by the SMART HMM (Figure 3.1D). The last helix of DP1 and the first helix of DP2 are both not covered by the HMM hits in the Sti1 sequence. Accordingly, we sought to understand to what degree the protein regions identified by this HMM actually reflect a single homologous family.
We performed structural alignments (Shindyalov and Bourne, 1998) (Figure 3.2A) across putative members of this family with experimentally solved structures (Sti1-DP1, Sti1-DP2, Tic40-STI1-II, and Rad23). Sti1-DP1 and Sti1-DP2 have clear homology indicated both by structural similarity (Figure 3.1B,C) and structure-guided sequence alignment using PROMALS3D (Pei et al., 2008) (Figure 3.1D). Tic40-STI1-II closely resembles Sti1-DP2 (Figure 3.2B). For Rad23, the orientation and register of the helices differs from the other domains. This can be visualized with respect to the substrate binding position where Rad23-STI1 forms the binding-groove with a rotated helix organization compared to the other STI1-domains (Figure 3.2B). In this orientation, the first helix of Rad23-STI1 aligns to the third helix of Sti1-DP2 and the fourth (last) helix of Rad23-STI1 occupies a position similar to the first two helices of Sti1-DP2. Based on alignments and structures Tic40-STI1 is clearly a member of the STI1-domain family whereas Rad23-STI1 may be erroneously annotated (Figure 3.2A).
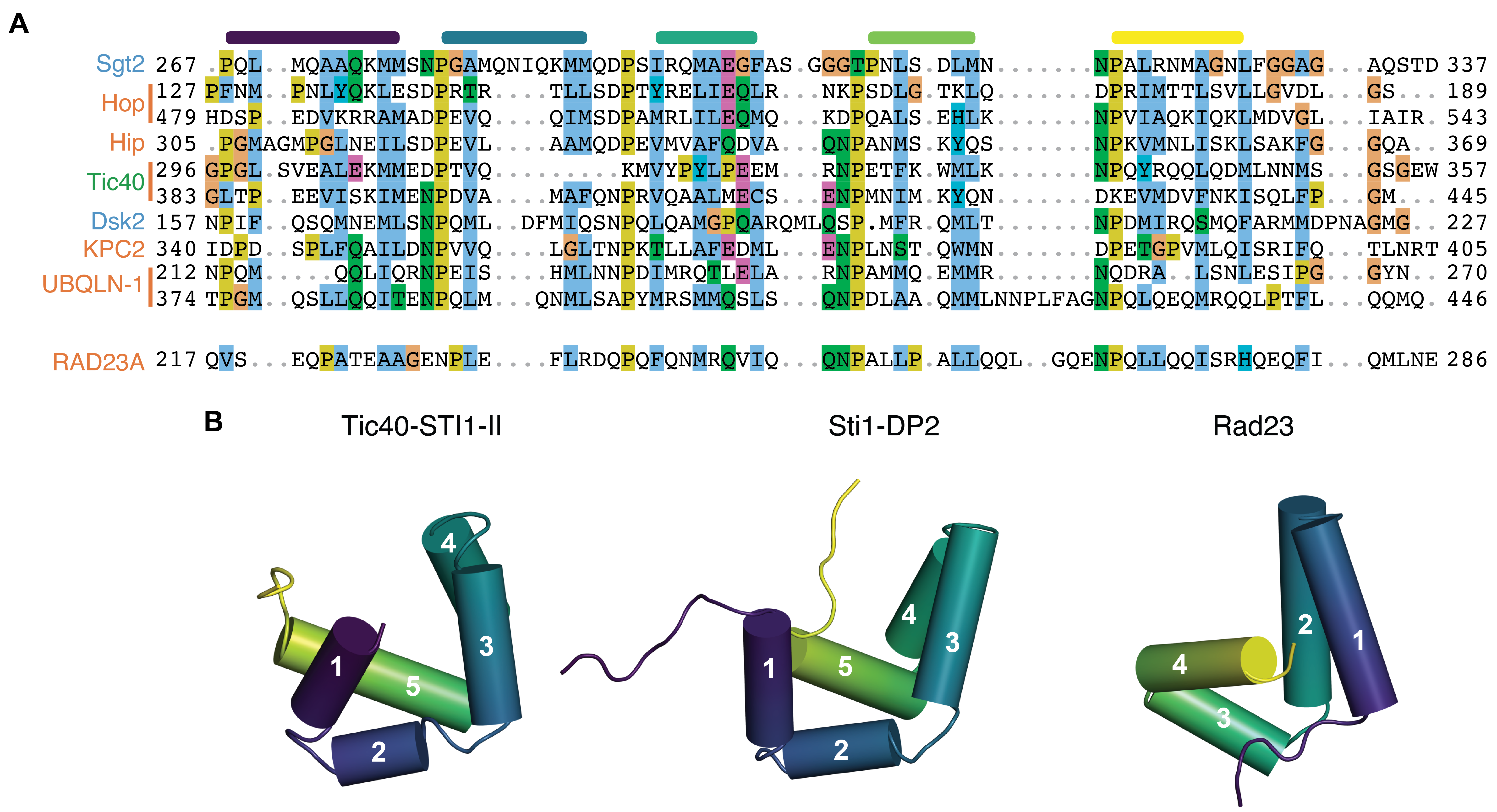
Despite its adequate utility, the SMART definition for STI1-domains can also lead to erroneous annotations of putative domains that have not been structurally characterized. Drawing the stretches of each protein with a hit from the SMART HMM alongside experimental or predicted helical regions, we can separate proper yet partial hits, e.g., Sti1-DP1 and Sti1-DP2, from potentially erroneous ones (Figure 3.3). An erroneous hit might originate from more than one consecutive hit to the HMM. UBQLN-1, -2, & -4 are predicted by the SMART HMM as having two pairs of abutting STI1-domains, for a total of four (Figure 3.3, Ubiquilin-1 I & II) (Letunic et al., 2015). In each case, the total length of each abutting hit was around 70 residues, not 100 which would be necessary for two STI1-domains considering the length of structurally determined STI1-domains. In addition, the secondary structure prediction within this region showed only seven helices (Figure 3.2A & Figure 3.2), sufficient for a single domain only. Thus, it is unlikely that there are two pairs of abutting domains.

As discussed earlier, the structure of the SMART identified STI1-domain in Rad23 differs from that of Sti1 DP domains and Tic40-STI1-II, again making it a possible erroneous annotation. The SMART HMM covers the same region as the Pfam XPC-binding HMM, but with a slightly lower score (27 vs 43); both align over a similar number of residues (40 vs 44). Given that the SMART HMM identifies the clear structural homolog Tic40-STI1-II with a similar score of 35, we cannot rule out Rad23 as a member of the STI1 containing family solely based on the SMART HMM score. Since the XPC-binding HMM uses Rad23-STI1 as part of the seed sequences, it could also be possible that the XPC-binding domain is a subfamily of STI1-domains. Although the Rad23 structure in this region appears distinct from other STI1-domain structures, it could be a member of this family based on its score and alignment to the SMART HMM. An HMM with higher specificity could more clearly delineate the difference between Rad23-STI1 and other STI1-domains, which we now aim to define.
3.3.2 A new definition for the STI1-domain
In light of these issues with the SMART definition for STI1-domains, we sought to generate an HMM that better defines the full-length of the STI1-domain and thereby more sensitively captures the full breadth of the STI1 family. First, we created an alignment of protein sequences that correspond to both structurally characterized STI1-domains and close homologs then aligned others with constraints from molecular models (see below) and/or secondary structure predictions. As expected, this multiple sequence alignment reveals a strong conservation of structural features (Figure 3.2A). The predicted helical regions, helices 1-5 (H1-H5), in several proteins align directly with the structurally determined helices of DP2 from Sti1 and Tic40-STI1-II. This includes complete conservation of helix breaking prolines and close alignment of hydrophobic residues defining amphipathic helices. The amphipathic nature of these helices appear important for client binding; experiments mutating the hydrophobic faces in these helices in SGTA to less hydrophobic alanine affected binding to tail-anchored protein substrates (Lin et al., 2021).
The resulting HMM clears up a number of the issues with the SMART HMM defining STI1-domains. Most identified STI1-domains align well between the two lists with the exception being Rad23 (Figure 3.2A). Along with other factors discussed later, this suggests Rad23 belongs to a class of STI1-like domains, which could include proteins like Ddi1 (Trempe et al., 2016). In the case of UBQLNs, the resulting HMM identifies the annotated abutting STI1-domains as a single STI1-domain (Figure 3.3).
Opposite to what was revealed in the UBQLNs, where a reduction in the number of identified STI1-domains was observed, a second N-terminal STI1-domain is identified by our HMM in Tic40 (Figure 3.3). Previously this region of Tic40 was suggested to be a TPR domain primarily on the basis of a binding to an anti-TPR1 antibody by western blot (Chou et al., 2003). While it is reasonable to suspect that Tic40 possesses a TPR domain since TPR domains precede STI1-domains in HOP, HIP, and SGTA, the TPR domain HMMs (El-Gebali et al., 2019) do not suggest a hit in this region. However, this region does produce a hit by our STI1 HMM. The anti-TPR1 antibody was generated against full-length rat TPR1 (Liu et al., 1999), and it is possible it lacks specificity for this plant TPR domain. We were unable to identify identical peptides longer than five residues between Tic40 and rat STI1 that could easily explain the cross reactivity. Due to the bioinformatic support for a STI1-domain in this region, we refer to it as Tic40-STI1-I and the structurally solved STI1-domain as Tic40-STI1-II.
3.3.3 STI1-domains are preceded by an N-terminal helix
Along with a curated set of STI1-domains, this new HMM reveals a conserved N-terminal sixth helix, hereafter referred to as Helix 0 (H0), which like H1-H5 is also amphipathic (Figure 3.4). As already noted, UBLQNs were mistakenly characterized as having four STI1-domains. The incorrect annotation was likely because three helices N-terminal to the domain were combined with the five helices of the STI1-domain, which resulted in eight contiguous helices recognized by SMART as two adjacent STI1-domains. The presence of N-terminal helices appears to be a general feature of STI1-domains. Based on secondary structure prediction and structures, it is clear, in most cases, that at least one helix precedes the STI1-domain, the exceptions being Sti1 DP2 and HIP (Figure 3.3). While the roles of the additional helices are not clear, H0 is well conserved within each protein and are also amphipathic in nature as the other helices (Figure 3.3, Figure 3.4).
3.3.4 Structural similarity between STI1-domains
With this new list of STI1-domains we inspected their predicted and structurally determined secondary structures. Broadly, these domains share several features including four to five amphipathic helices, as annotated (Figure 3.2A). For STI1-domains that have been structurally characterized (DP1, DP2, Tic40-STI1-II), the helices assemble into a tertiary structure that resembles a helical-hand forming a hydrophobic groove (Figure 3.5A-C) and are characterized by structural flexibility (Lin et al., 2021). Though no structures of a STI1-domain from a co-chaperone exist with a client occupying the hydrophobic groove, it presents an appealing pocket for the binding site of hydrophobic segments. The flexibility may contribute to the ability of these domains to specifically bind and then release substrates as part of their functional role.
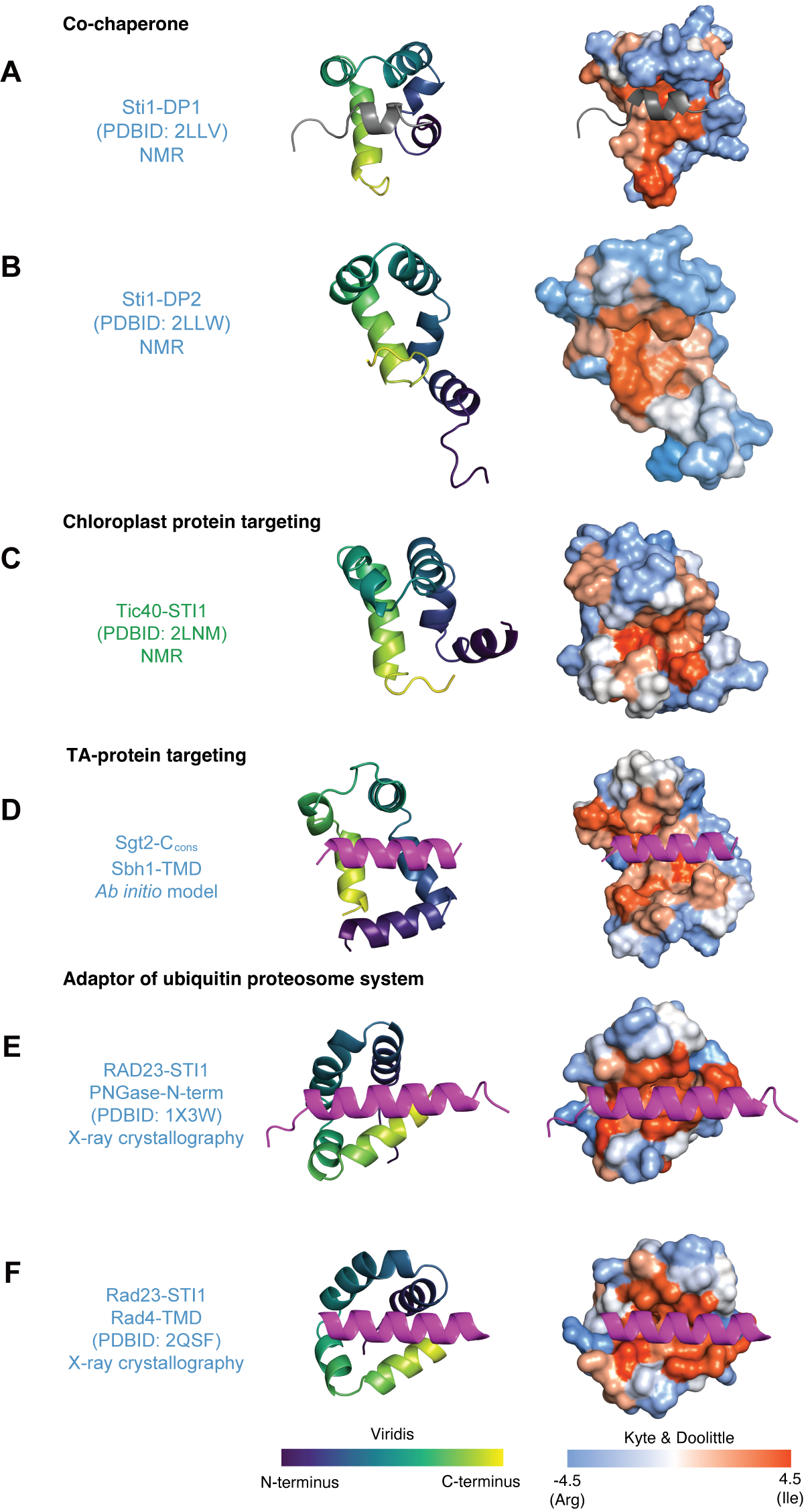
DP1 and DP2 were the first STI1-domains to be structurally characterized. The two structures have five amphipathic alpha helices arranged like a cupped hand presenting a hydrophobic groove. In the structure of DP1 the hydrophobic hand is occupied by H0, possibly mimicking client binding (grey helix Figure 3.5A). Both structures of DP1 and DP2 were solved using NMR. When comparing the states from the models for each domain, DP2 appears to be more flexible than DP1, with the N-terminal H0 in DP1 likely stabilizing its core region (Schmid et al., 2012).
Tic40 contains another structurally characterized STI1-domain in the absence of a client occupying the groove. Like DP1 and DP2, Tic40-STI1-II consists of five alpha helices that arrange into a similar helical hand with a hydrophobic groove (Figure 3.5C). Tic40 is predicted to have an H0 that was not included in the determined structure. Other STI1-domains have remained resistant to structure determination.
Alternative structural methods have been used to characterize other STI1-domains. One domain in particular is the C-terminal domain of fungal homolog of SGTA, Sgt2, which remains recalcitrant to experimental structural determination. Ab initio molecular modeling of Sgt2-C followed by experimental validation of residue-pair distances further suggests that the domain is part of the STI1 family (Lin et al., 2021) (Figure 3.5D). Residues of a conserved region resolve a potential binding interface for a helical hydrophobic substrate. Outside this region they adopt varied conformations consistent with expected high flexibility. The working model contains a potential TA client binding site – a hydrophobic groove formed by the amphipathic helices. The groove is approximately 15 Å long, 12 Å wide, and 10 Å deep, which is sufficient to accommodate three helical turns of an alpha-helix, ~11 amino acids. Like the NMR structures of other STI1-domains found in co-chaperones, the ab initio model of Sgt2-C resembles the general STI1-domain structure.
For the STI1-domain containing AUPS proteins, no experimental or ab initio structures currently exist. To predict structures of these STI1-domains including those in the UBQLNs we employed the Robetta transform-restrained (TR) tool, a state-of-the-art structure prediction method (Yang et al., 2020) where a deep neural network predicts pairwise residue distances and angles followed by energy minimization. Distinct from template- or fragment-based approaches, Robetta TR generates de novo structures from restraints where structures are not explicitly used. As validation for our domains, we first compare the prediction of multiple STI1-domains by Robetta TR versus the experimentally derived structure. Providing the full-length sequences from ScSti1 and AtTic40, Robetta TR provides a model of the full-length protein (Supplementary Figure 3.1A). We isolated the STI1-domains from each and compare them to the structures solved by NMR (Figure 3.6A, Supplementary Figure 3.1). For the top predicted models, DP1, DP2, and Tic40-STI1-II have five helices that assemble into a helical hand (Figure 3.6A, Supplementary Figure 3.1B,C). These predictions are in close agreement with the NMR derived structures, with the last five helices of the prediction overlaying with the five alpha helices in the NMR structure – supporting that the prediction method can provide data broadly on STI1-domains (Figure 3.6A, Supplementary Figure 3.1B,C).
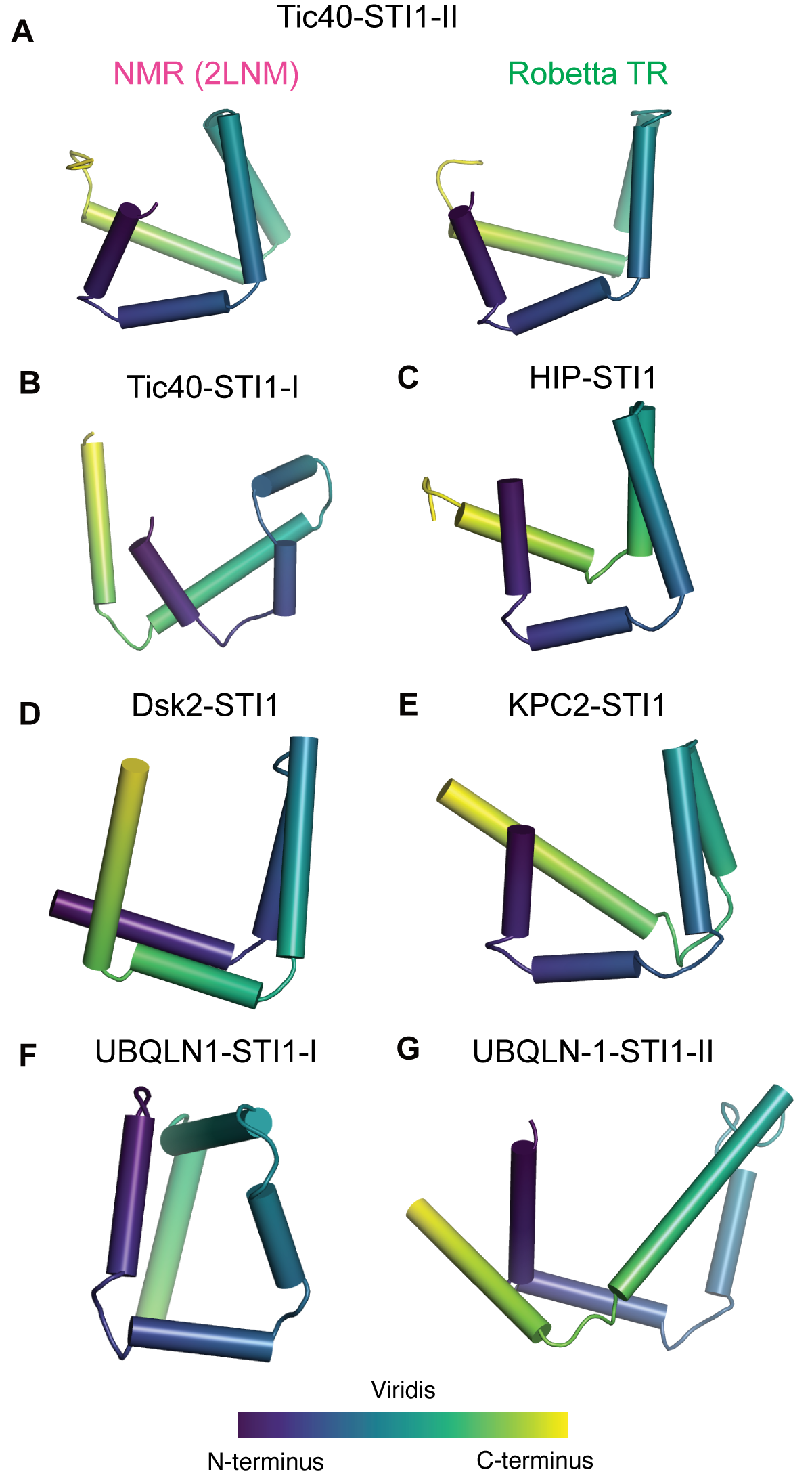
We proceeded to predict the structures of uncharacterized STI1-domains in other co-chaperones. The predicted structure for Tic40-STI1-I has a similar fold to other STI1-domains, the five alpha-helical hand, supporting the new domain definition (Figure 3.6B). This model further reduces the likelihood that this region contains a TPR domain that are structurally distinct alpha-solenoids. The STI1-domain from human HIP (Figure 3.6C), the last of the uncharacterized co-chaperone STI1-domains, is similar to the experimentally determined Tic40-STI1-II (Figure 3.6A), DP2, and DP1 (Figure 3.5A,B). Across all co-chaperones we observe five helices coming together to form an alpha helical hand.
We next predicted the structures of the uncharacterized STI1-domains in the AUPS family (Dsk2, KPC2, and UBQLNs) (Figure 3.6D-G). Like HIP, the predicted structure of KPC2 is consistent with the experimentally determined structures of STI1-domains from the co-chaperones. The predicted structure of the Dsk2 and UBQLN-1 also display a variation of the STI1 helical hand observed in the solved structures (Figure 3.6D,F,G). A helical groove compatible for binding a hydrophobic alpha helix is formed by five helices in each of these cases. Dsk2-STI1 (Figure 3.6D) and the second STI1-domain of UBQLN-1 (UBQLN-1-STI1-II) differs from the co-chaperone STI1-domains as their helices form the groove in the reverse order (Figure 3.6G). Unlike the prediction of UBQLN-1-STI1-II, the first STI1-domain (UBQLN-1-STI1-I) forms a nearly enclosed groove, which could accommodate client binding with some rearrangement (Figure 3.6F). These predictions are models for what these STI1-domains could look like and in both the co-chaperones and AUPS these models suggest that these STI1-domains can form helical hands to accommodate client binding. UBQLNs (UBQLN 1-4) are a particularly important focus of research and experimental work to test these predictions will be broadly useful (Deng et al., 2011; Itakura et al., 2016; Marín, 2014; Şentürk et al., 2019; Subudhi and Shorter, 2018).
The structure of Rad23-STI1 bound to substrate supports the exclusion of Rad23 from the STI1-domain containing family of proteins (Figure 3.5D,E). While several structures of complexes of Rad23-STI1 bound to amphipathic clients show in each that the client-helix binds via a hydrophobic groove, the domain architecture differs from those determined in the co-chaperones and the predicted structures of other STI1-domain containing proteins (Figure 3.2C, Figure 3.6B-G). Despite Rad23-STI1 being a helical bundle that binds clients similar to the co-chaperones, the absence of a fifth helix supports that Rad23 does not contain a STI1-domain, but is instead a STI1-like domain which also utilizes a hydrophobic groove. It has been observed that the first three helices of the Rad23 XCP domain are structurally similar to the first three helices of the N-terminal domain of the Helical Domain (HDDnt) from the DNA damage inducible 1 protein (Ddi1), but the fourth helix deviates and goes in a different direction (Trempe et al., 2016). Ddi1 differs from other shuttle proteins because of its proteolytic role and interacting partners (Zientara-Rytter and Subramani, 2019). Like the shuttle proteins described above, Ddi1 contains a UBA and UBL domain, but the UBA domain has been lost in mammalian homologs. Ddi1 also contains a retrovirus protease (RVP) fold domain. The UBL domain of Ddi1 has an unusual binding preference, unlike the domain in Rad23 or Dsk2, it does not interact with its UBA domain or Rpn10 and interacts weakly with Rpn1. It has been suggested that Ddi1 may assist Rad23 or Dsk2 instead of acting as a shuttle factor on its own.
Due to its homology with the XCP domain of Rad23, which has been implicated through protein-protein interactions, HDDnt may play a similar role (Trempe et al., 2016). It has also been observed that HDDnt has a similar structure to other DNA binding domains suggesting HDDnt may bind directly to DNA. With structural similarities to various domains, it is reasonable to think Rad23 and Ddi1 both contain STI1-like domains.
3.3.5 Similarity in the domain structures of STI1 proteins
When examining the predicted secondary structure of the entirety of STI1-domain containing proteins several common characteristics became clear. Dual STI1-domains are present in both the co-chaperones and AUPS (Figure 3.7). Within the co-chaperones, two distinct groups emerge — ones that possess two STI1-domains (HOP, Tic40) and those that possess a dimerization domain (SGTA, HIP). As stated previously, HOP contains two STI1-domains separated by multiple TPR domains with both required for efficient client transfer (Schmid et al., 2012). It has been speculated that DP1 and the first TPR domain (TPR1) act as an intermediate in the shuttling of a client from Hsp70 to the TPR2A&B and DP2-bound Hsp90 (Biebl and Buchner, 2019; Kirschke et al., 2014; Lott et al., 2020; Reidy et al., 2018; Röhl et al., 2015, 2013; Schmid et al., 2012; Schopf et al., 2017).
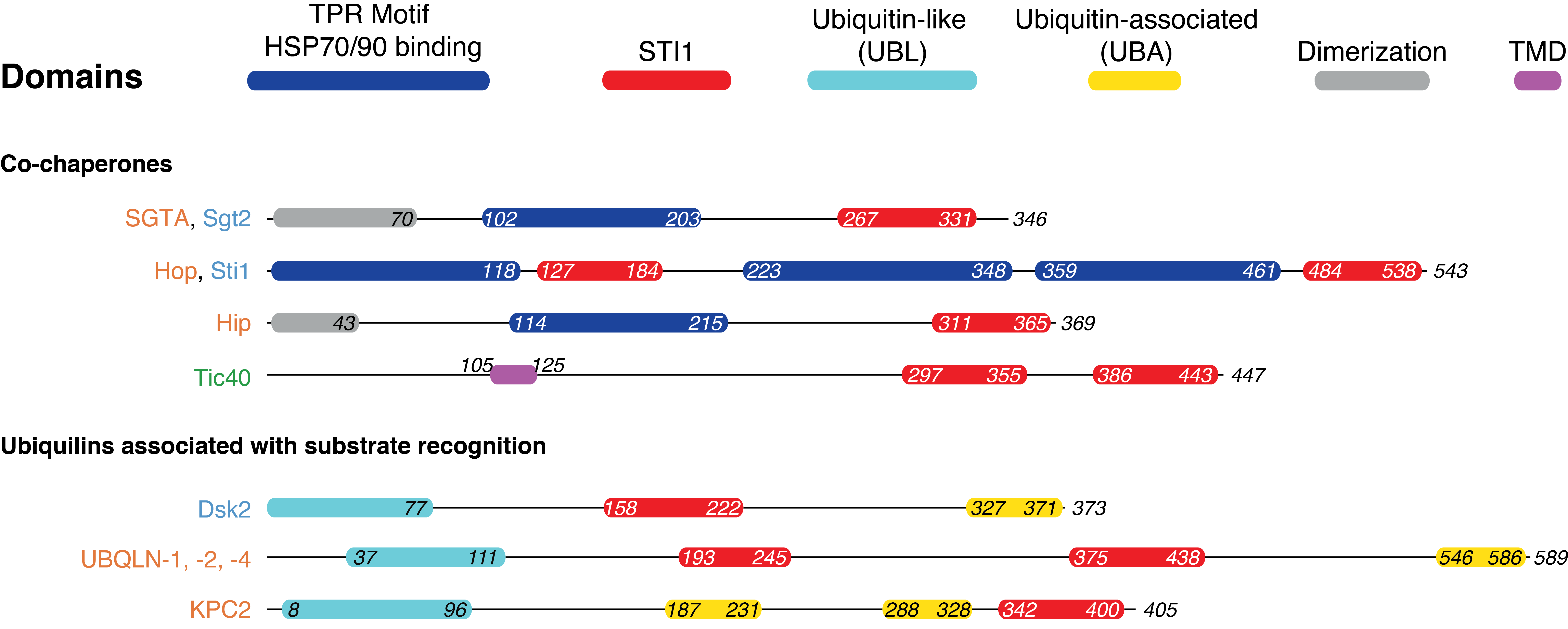
Unlike HOP, Tic40 contains two abutting STI1-domains. Given our recent identification of the first STI1-domain, its function has yet to be determined. Found in the chloroplast inner membrane with the STI1-domains in the stroma, the C-terminal domain can be replaced with the STI1-domain from HIP without loss-of-function (Bédard et al., 2017). In HIP the STI1-domain interacts with the leucine-rich chemokine receptor (Fan et al., 2002), the previously proposed TPR domain, now STI1-I, of Tic40 interacts with the leucine-rich mature region of Tic110 (Bédard et al., 2007).
The homodimerization of HIP and SGTA, each containing a single STI1-domain in the monomer, results in a complex with two STI1-domains. Small angle X-ray scattering (SAXS) data has revealed that for both proteins in solution, the dimers (Chartron et al., 2011) form elongated, flexible complexes (Li et al., 2013). The elongated form would put maximal distance between the two STI1-domains which are found opposite of the dimerization domains. The fact that the STI1-domain from HIP can functionally replace a STI1-domain from Tic40, both in pairs, but one through dimerization domain and the other encoded in the monomer, suggests the STI1-domains have similar overall functions, the significance of these pairs in the co-chaperones is still unclear.
The presence of a pair of STI1-domains is also observed in UBQLNs. As discussed earlier, UBLQNs contain two STI1-domains and not four as previously thought. This clarification relates UBQLNs to the co-chaperones HOP and Tic40, where each have a STI1-domain pair encoded in their monomer, while still separating them from other AUPS family members which contain a single STI1-domain (Figure 3.7). The observation of a pair of STI1-domains in UBLQNs and the previously identified active roles in protein targeting and degradation (Itakura et al., 2016) set these proteins apart from the other AUPS family members. The previously defined M domain of UBQLN-1 contains both identified STI1-domains and is responsible for its ability to shield TMDs of mitochondrial membrane proteins from the cytosol and to deliver them for degradation (Itakura et al., 2016). Identifying pairs of STI1-domains in both co-chaperones and UBLQNs, proteins with a known role in protecting TMDs in the cytosol through their STI1-domains, suggests these pairs aid in this role.
While a pair of STI1-domains is a found in both AUPS and co-chaperone proteins, an HSP-binding TPR domain preceding the STI1-domain(s) connected by a flexible linker is exclusively observed in the co-chaperones (Figure 3.7). These TPR domains have been shown to aid in client hand-off in these proteins. The multiple TPR domains in HOP are used to coordinate simultaneous binding of Hsp70 and Hsp90, facilitating client transfer between the two chaperones (Scheufler et al., 2000; Schmid et al., 2012; Zeytuni and Zarivach, 2012). In contrast, HIP contains a TPR domain that only interacts with Hsp70. Additionally in Sgt2, the TPR domain increases the efficiency of capture of TA clients by coordinating with a client bound Hsp70 homolog, Ssa1 (Cho and Shan, 2018). While HOP has two TPR domains within a monomer, both SGTA and HIP link two TPR and STI1-domains by forming stable dimers via N-terminal dimerization domains(Coto et al., 2018). For the SGTA and HIP homodimers, a cooperative role between the two copies of each TPR- and STI1-domain remains a possibility.
Differing from the other co-chaperones, the relatively more distant chloroplast Tic40 has its own domain architecture. Previously, the N-terminal STI1-domain was annotated as a TPR domain but, as discussed earlier, bioinformatics and computational models counter this claim. The rest of the protein lacks a clear TPR domain and has an N-terminal TMD. How Tic40 fits mechanistically into this group of co-chaperones is less clear due to it missing a TPR domain and being membrane bound.
3.3.6 Amino acid distribution in STI1-domains
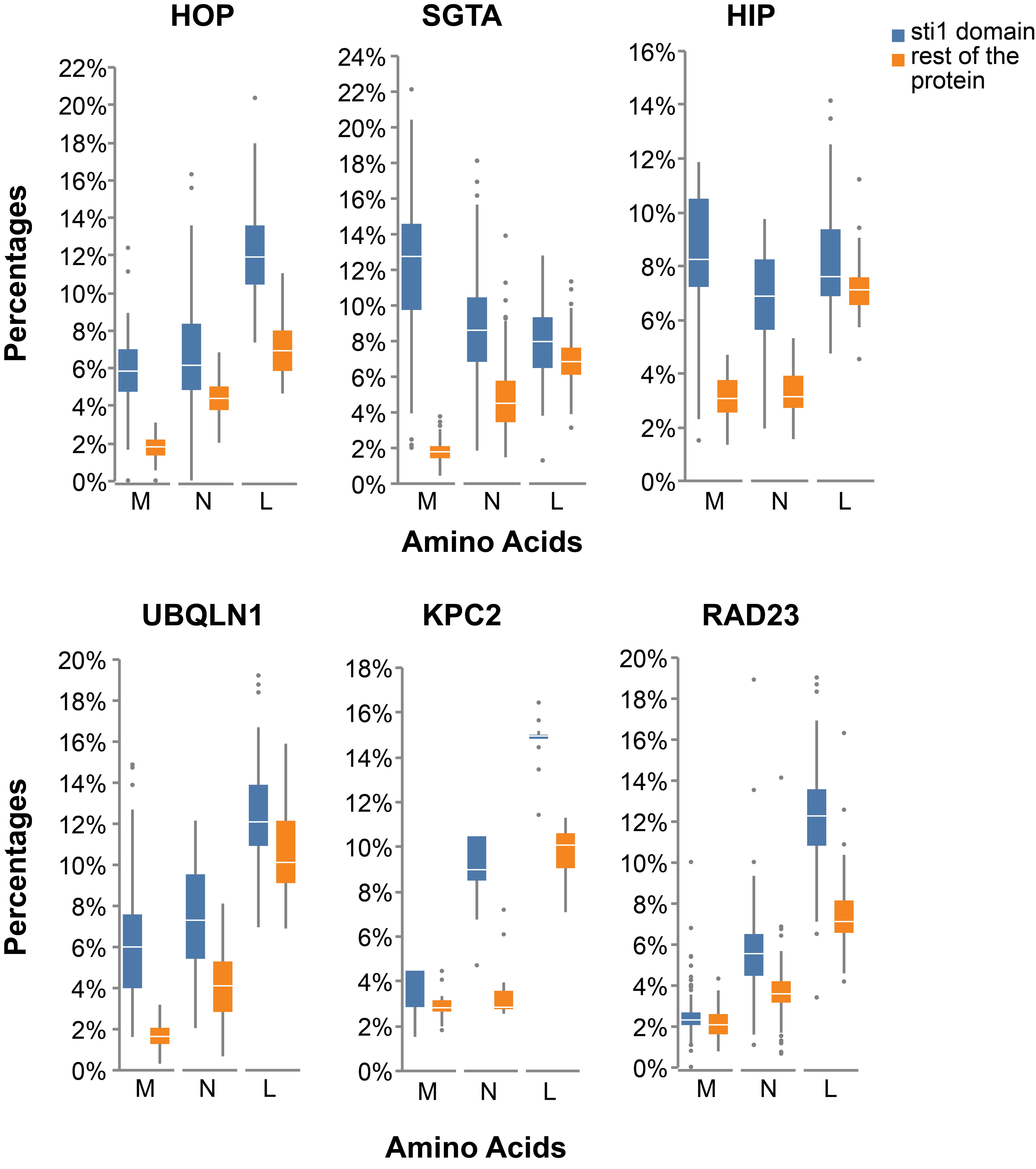
STI1-domains were initially described as DP domains due to two repeats of a DPEV motif in HIP (Prapapanich et al., 1998) and a DPEV and DPAM motif in HOP (Chen and Smith, 1998). From the solved structures (Figure 3.5) and predicted secondary structure (Figure 3.3), we see this motif localizing to the N-terminus of helices likely acting as a cap. Aspartate and threonine most frequently occur at the cap of a helix often followed by either a glutamine or proline (Aurora and Rose, 1998). The role for this motif where found is likely as a stabilizing N-cap accounting for its conservation. When analyzed broadly, a repeat DP motif is not observed in the majority of STI1-domains, it not even found in all HOP homologs.
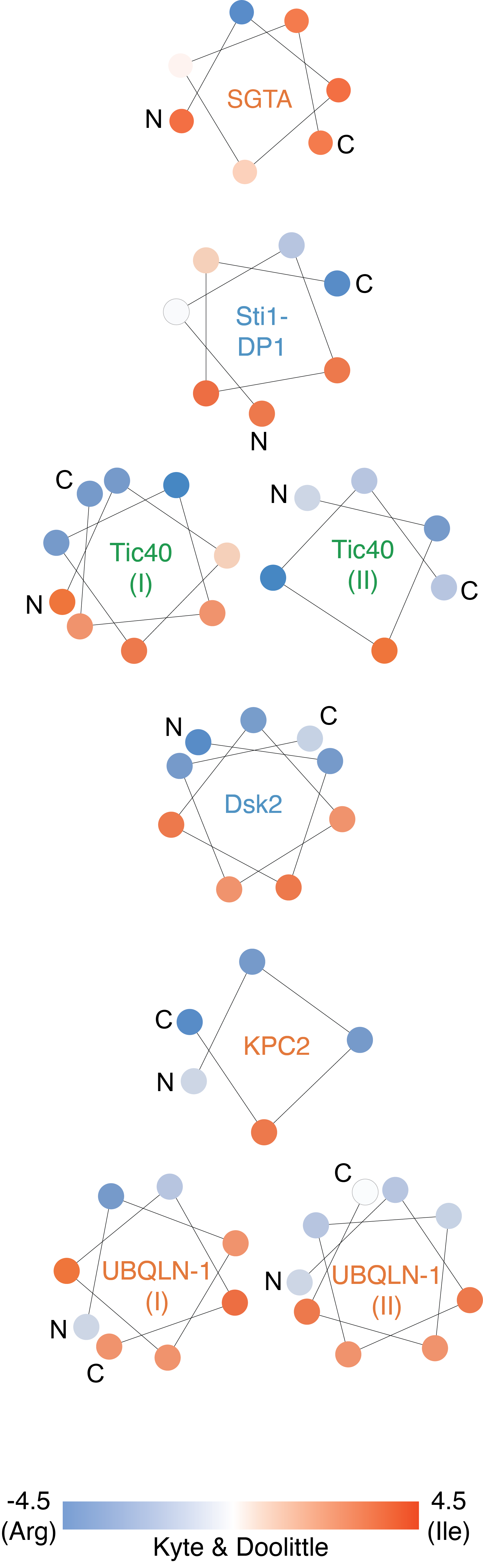
We were interested if there were common residues overrepresented in STI1-domains, considering that in some cases these domains have been referred to as methionine-rich (Itakura et al., 2016; Lin et al., 2021). To quantify this, we began with compiling a list of homologs for HOP, SGTA, HIP, UBQLN-1, and KPC2 using EnsemblGenomes (Burri and Lithgow, 2004) and then calculated the distribution of amino acids within the STI1-domain. As for Dsk2 homologs, only two hits were found after searching EnsemblGenomes, therefore we omitted Dsk2 from this analysis. Only an overrepresentation of methionine, asparagine, and leucine is observed across all five protein groups (Figure 3.8). An overrepresentation of methionine has also been observed in other hydrophobic segment binding domains such as the M domain of the signal recognition particle (SRP) (Bernstein et al., 1989; Zopf et al., 1990) and Get3 (Mateja et al., 2009; Suloway et al., 2009). As discussed previously, Rad23 likely does not contain a STI1-domain. We applied the same analysis of the amino acid distribution in the previous annotated STI1-domain of Rad23 and found that, unlike verified STI1-domains, methionine is not overrepresented (Figure 3.8). Overall, this analysis reveals that over representation of methionine, asparagine, and leucine is a feature of STI1-domains.
3.4 Discussion
STI1-domains have been predicted in a number of proteins essential for protein biogenesis. Here we explicate a definition for STI1-domains and curate a list of STI1-domain containing proteins through structure-based sequence alignments, validating some previously predicted domains as well as identifying new ones. Solved structures and computational models reveal STI1-domains consist of five to six helices organized into a helical hand with a hydrophobic groove. Upon close inspection, STI1-domain containing proteins can be classified into two families—co-chaperones or AUPS — with several common features noted. Overall, this work presents the first in-depth examination of STI1-domains and the essential proteins for where they are found.
Previously, STI1-domains were identified by the SMART database, which lead to the identification of erroneous hits and omissions for the family. Our revised HMM that encompasses a minimal five helix region helps uncover and clarify the full breadth of STI1-domains. As a result, this new definition revealed new STI1-domains and corrections to previous identifications. For example, the annotations of four abutting STI1-domains in UBQLNs are actually a set of two STI1-domains and the annotated TPR domain in the co-chaperone Tic40 is more likely a STI1-domain. Furthermore, it is now clear that Rad23 does not contain a STI1-domain, but has a distinct helical hand formed of only four helices.
This revised list was evaluated to determine common structural features. The overall five amphipathic helices forming a flexible helical hand are seen in the structures of DP2 and Tic40-STI1-II (Schmid et al., 2012). Differing from the structure of the DP2, the DP1 structure contains a sixth helix (H0) that resides in the groove (Figure 3.1B, Figure 3.5). With our new HMM, an H0 was identified preceding most STI1-domains and its role is yet to be determined (Figure 3.3). Due to the flexibility of STI1-domains, one possibility is that H0 fills the groove as seen in the DP1 structure (Guna and Hegde, 2018). In this model the hydrophobic residues in H0 would dock in the groove of the helical hand stabilizing the domain in the absence of client. H0 would then be displaced by an appropriate client.
Outside of the STI1-domains, several common features surface in these STI1-domain containing proteins. A Distinct feature of the co-chaperone family are the TPR domains. We previously discussed the various TPR domains identified in Sgt2, HOP, and HIP, noting sequence features that define specificity in interacting partners (Chartron et al., 2011). TPR domains consist of multiple repeats of 34 amino acids in a helix-turn-helix fold, with anti-parallel alpha-helices. Differences in the binding pocket of TPR domains allow for selectivity of a diverse set of chaperone partners. In the case of SGTA, the TPR-domain works together with the STI1-domain to coordinate client binding. Ssa1 carrying STI1-domain clients interact directly with the TPR domains allowing for client capture by the STI1-domains. Here we demonstrate that the suggested TPR domain of Tic40 is more likely a STI1-domain based on its higher score against the STI1 HMM vs the TPR HMM. This adjustment to the domain structure within Tic40 may suggest that Tic40 does not interact with HSPs to capture clients as seen for the other co-chaperones.
Pairs of STI1-domains are found in both co-chaperones and the AUPS family, either encoded in a single monomer or joined through a dimerization region. While the co-chaperones identified in this paper contain STI1-domains in pairs, for the AUPS family this is only true for UBQLNs. Of the AUPS family members, UBQLNs are the only ones so far shown to play a direct role in preventing client aggregation in the cytosol and facilitating the degradation of mitochondrial membrane proteins that fail to insert into the mitochondrial membrane (Itakura et al., 2016). These roles in both protein targeting and degradation are similar to those of SGTA — handing off TA clients to chaperones in the GET pathway for insertion and handing off mislocalized proteins to Bag6 for degradation (Leznicki and High, 2012). The pair of STI1-domains lie in the identified client binding domain of UBQLN-1. Perhaps due to similar roles, these domains in UBQLNs function similarly to the STI1-domains in SGTA. The details of how STI1-domain pairs affect function and if they interact with one-another are important areas for future study.
How might pairs of STI1-domains cooperate for client specificity and selection? Conceptually, a pair of STI1-domains may simultaneously bind the same client TMD in the case of UBQLNs and SGTA. We have shown previously that a single STI1-domain from Sgt2 can bind to a minimum of 11 amino acids in a client (Lin et al., 2021). Conceivably, one model is that the two STI1-domains in the Sgt2 dimer simultaneously bind a single client — binding side-by-side on a client TMD that averages 20aa. This would require that these domains come close together altering the overall architecture. A related model is that the pair of STI1-domains could cooperate to increase the apparent affinity for a client TMD by increasing the local concentration of the binding domain. The simplest model is that each STI1-domain binds a separate client either to increase client load (two clients per monomer/dimer instead of one) or there is a necessity to bind two clients at once. On the other hand, it is also possible that two STI1-domains are necessary for different functions, as proposed previously in the case of HOP by Schmid and colleagues where glucocorticoid receptor activation cannot by rescued by replacing DP2 with DP1. It is worth noting that low resolution structural studies have suggested that dimeric Sgt2 and HIP position their STI1-domains on opposite ends of a dimer molecule in the absence of client (Chartron et al., 2011; Coto et al., 2018). Still, given the noted flexibility in these proteins (Lin et al., 2021; Schmid et al., 2012), the possibility of cooperation remains, with the molecular details an open question.
Flexibility is a common motif seen within STI1-domains and the proteins where they are identified. NMR studies of STI1-domains have suggested that these domains are flexible. We consider this flexibility a feature of these helical-hands for reversible and specific binding of a variety of clients. But what is the benefit of the flexible helical-hand structure for hydrophobic helix binding? While it remains an open question, it is notable that evolution has settled on similar simple solutions to the complex problem of specific but temporary binding of hydrophobic helices. For all of the domains with experimentally determined structures, the flexible helical-hands provide an extensive hydrophobic surface to capture the client-helix. Required to only engage temporarily, the flexibility of the helical hand could offset the favorability of the domain to bind a hydrophobic client, allowing the client to be released. This would account for the favorable transfer seen from Sgt2 (Cho and Shan, 2018) and SGTA (Shao et al., 2017) to downstream components.
This work provides a comprehensive HMM to define and identify STI1-domains in proteins and recognizes common features observed within the domains themselves and across STI1-domain containing proteins. These patterns leave open questions that have yet to be determined. What is the role of flexibility within STI1-domains and STI1-domain containing proteins? Does helix zero act as a stand in for clients in their absence or does it have a role as a lid? What is the benefit of having two STI1-domains in the co-chaperones? Do UBQLNs have a larger role than other AUPS family members and do their two STI1-domains contribute to this different role? There is much still to be understood about the underlying mechanisms that result in specificity and client handoff of STI1-domains. This comprehensive list of STI1-domains provides a coherent starting point.
3.5 Material and Methods
3.5.1 Molecular visualization
All STI1-domains with experimentally determined structures were retrieved from the RCSB PDB. Ab initio structure prediction was employed for other STI1-domains using RobettaTR (transform restrained) (Yang et al., 2020) with the full-length protein sequences of each protein, with the specific STI1 region of interest visualized. Images were rendered using PyMOL 2.4 (www.pymol.org) with a viridis coloring scheme (Saladi et al., 2020). Helical wheel diagrams were rendered in R using a fork (https://github.com/smsaladi/heliquest) of the HELIQUEST source code (Gautier et al., 2008).
3.5.2 Sequence analyses
Alignments of Sti1 (DP1/DP2) and STI1-domains were created by pulling all unique domain structures with annotated STI1-domains from Uniprot. Sequences were clustered at 50% similarity present, with the human, yeast, and A. thaliana preferred and then aligned with PROMALS3D (Pei et al., 2008) along with all experimentally determined structures of STI1-domains. PROMALS3D provides a way of integrating a variety of costs into the alignment procedure, including 3D structure, secondary structure predictions, and known homologous positions. The human, yeast, and A. thaliana homologs were selected from this alignment for display. An HMM for the STI1 domain was generated using HMMER v3.3.1. Alignments were visualized using Jalview (Waterhouse et al., 2009). Secondary structure where indicated is calculated using DSSP (Kabsch and Sander, 1983) on experimentally determined or predicted structures (Yang et al., 2020).
3.5.3 Amino acid composition of STI1-domains
Homologs of ScSti1, HsHOP, ScSgt2, HsSGTA, HsHIP, ScDsk2, ScKPC2, HsUBQLN-1, and HsRad23A were compiled from EnsemblGenomes (Howe et al., 2020) and filtered for redundancy at 70% sequence identity using the CD-HIT Suite (Li and Godzik, 2006). Sequences were then aligned using MAFTT (Katoh and Standley, 2013). The STI1-domain(s) in each sequence were identified by alignment to the known STI1-domains of HsHOP and HsSGTA to yield two segments: the STI1-domain and the non-STI1-domain region, i.e., the “rest” of the protein. For each segment of each protein the percentage of all individual amino acids was calculated. Significance was determined by permutation testing, comparing the difference between the first quartile of an amino acid’s percentage between each segment, i.e., within vs outside of the STI1-domain.
3.5.4 Data Availability
Our refined HMM for the STI1-domain (HMMER format), alignment shown in Figure 3.2 (FASTA format), and each of the structural models (PDB format) are included as supplementary data. Structural models should be used with caution.
3.6 Supplementary Figure