| Pfam ID | Pfam Name | Expression Trials | Positive Outcomes | Negative Outcomes | AUC (95% CI) |
|---|---|---|---|---|---|
| PF00359.19 | PTS_EIIA_2 | 14 | 3 | 11 | 1.00 (1.00-1.00) |
| PF02656.12 | DUF202 | 10 | 9 | 1 | 1.00 (1.00-1.00) |
| PF03186.10 | CobD_Cbib | 11 | 1 | 10 | 1.00 (1.00-1.00) |
| PF03176.12 | MMPL | 105 | 1 | 104 | 0.99 (0.99-0.99) |
| PF02683.12 | DsbD | 18 | 1 | 17 | 0.97 (0.97-0.97) |
| PF02355.13 | SecD_SecF | 33 | 1 | 32 | 0.97 (0.97-0.97) |
| PF12822.4 | DUF3816 | 11 | 4 | 7 | 0.96 (0.87-1.00) |
| PF00662.17 | Proton_antipo_N | 120 | 2 | 118 | 0.94 (0.87-1.00) |
| PF04247.9 | SirB | 16 | 1 | 15 | 0.93 (0.93-0.93) |
| PF06127.8 | DUF962 | 11 | 6 | 5 | 0.93 (0.78-1.00) |
| PF11742.5 | DUF3302 | 15 | 1 | 14 | 0.93 (0.93-0.93) |
| PF01610.14 | DDE_Tnp_ISL3 | 14 | 2 | 12 | 0.92 (0.73-1.00) |
| PF07290.8 | DUF1449 | 11 | 1 | 10 | 0.90 (0.90-0.90) |
| PF02659.12 | Mntp | 29 | 2 | 27 | 0.89 (0.76-1.00) |
| PF04217.10 | DUF412 | 12 | 5 | 7 | 0.89 (0.65-1.00) |
| PF05425.10 | CopD | 20 | 3 | 17 | 0.88 (0.73-1.00) |
| PF02535.19 | Zip | 34 | 1 | 33 | 0.86 (0.86-0.86) |
| PF02667.11 | SCFA_trans | 19 | 7 | 12 | 0.86 (0.68-1.00) |
| PF01810.15 | LysE | 41 | 2 | 39 | 0.83 (0.59-1.00) |
| PF06930.9 | DUF1282 | 12 | 5 | 7 | 0.83 (0.57-1.00) |
| PF03595.14 | SLAC1 | 76 | 19 | 57 | 0.81 (0.67-0.94) |
| PF04172.13 | LrgB | 63 | 4 | 59 | 0.79 (0.57-1.00) |
| PF03601.11 | Cons_hypoth698 | 52 | 9 | 43 | 0.78 (0.64-0.92) |
| PF02537.12 | CRCB | 81 | 20 | 61 | 0.77 (0.66-0.88) |
| PF01757.19 | Acyl_transf_3 | 23 | 6 | 17 | 0.76 (0.53-1.00) |
| PF02378.15 | PTS_EIIC | 82 | 15 | 67 | 0.76 (0.62-0.90) |
| PF03929.13 | PepSY_TM | 25 | 2 | 23 | 0.76 (0.36-1.00) |
| PF00999.18 | Na_H_Exchanger | 398 | 21 | 377 | 0.76 (0.66-0.86) |
| PF00535.23 | Glycos_transf_2 | 282 | 62 | 220 | 0.76 (0.70-0.82) |
| PF03605.11 | DcuA_DcuB | 28 | 2 | 26 | 0.75 (0.32-1.00) |
| PF02690.12 | Na_Pi_cotrans | 64 | 9 | 55 | 0.75 (0.54-0.95) |
| PF01699.21 | Na_Ca_ex | 75 | 4 | 71 | 0.74 (0.58-0.90) |
| PF03390.12 | 2HCT | 18 | 3 | 15 | 0.73 (0.37-1.00) |
| PF00950.14 | ABC-3 | 33 | 7 | 26 | 0.73 (0.51-0.94) |
| PF06762.11 | LMF1 | 17 | 1 | 16 | 0.72 (0.72-0.72) |
| PF03741.13 | TerC | 19 | 4 | 15 | 0.72 (0.42-1.00) |
| PF02632.11 | BioY | 47 | 15 | 32 | 0.71 (0.54-0.88) |
| PF06738.9 | ThrE | 26 | 2 | 24 | 0.71 (0.42-1.00) |
| PF11911.5 | DUF3429 | 36 | 12 | 24 | 0.70 (0.53-0.88) |
| PF01226.14 | Form_Nir_trans | 13 | 8 | 5 | 0.70 (0.38-1.00) |
| PF09586.7 | YfhO | 6 | 1 | 5 | 0.70 (0.70-0.70) |
| PF03739.11 | YjgP_YjgQ | 214 | 17 | 197 | 0.70 (0.56-0.84) |
| PF05656.11 | DUF805 | 100 | 21 | 79 | 0.69 (0.56-0.81) |
| PF01566.15 | Nramp | 129 | 21 | 108 | 0.69 (0.55-0.82) |
| PF05977.10 | MFS_3 | 12 | 3 | 9 | 0.69 (0.37-1.00) |
| PF03023.11 | MVIN | 67 | 7 | 60 | 0.68 (0.46-0.90) |
| PF02674.13 | Colicin_V | 32 | 4 | 28 | 0.68 (0.47-0.89) |
| PF03009.14 | GDPD | 46 | 1 | 45 | 0.68 (0.68-0.68) |
| PF10110.6 | GPDPase_memb | 46 | 1 | 45 | 0.68 (0.68-0.68) |
| PF01545.18 | Cation_efflux | 21 | 5 | 16 | 0.68 (0.32-1.00) |
| PF07854.9 | DUF1646 | 7 | 1 | 6 | 0.67 (0.67-0.67) |
| PF01594.13 | UPF0118 | 436 | 137 | 299 | 0.67 (0.61-0.72) |
| PF06541.8 | ABC_trans_CmpB | 33 | 2 | 31 | 0.66 (0.51-0.81) |
| PF12698.4 | ABC2_membrane_3 | 248 | 13 | 235 | 0.65 (0.46-0.85) |
| PF01578.17 | Cytochrom_C_asm | 53 | 5 | 48 | 0.65 (0.39-0.92) |
| PF00520.28 | Ion_trans | 17 | 4 | 13 | 0.65 (0.35-0.95) |
| PF00230.17 | MIP | 83 | 35 | 48 | 0.65 (0.53-0.77) |
| PF04608.10 | PgpA | 31 | 11 | 20 | 0.65 (0.43-0.87) |
| PF03062.16 | MBOAT | 78 | 5 | 73 | 0.65 (0.36-0.94) |
| PF01569.18 | PAP2 | 8 | 1 | 7 | 0.64 (0.64-0.64) |
| PF03788.11 | LrgA | 55 | 3 | 52 | 0.64 (0.36-0.92) |
| PF03824.13 | NicO | 15 | 3 | 12 | 0.64 (0.26-1.00) |
| PF03222.10 | Trp_Tyr_perm | 13 | 5 | 8 | 0.64 (0.35-0.92) |
| PF01040.15 | UbiA | 212 | 29 | 183 | 0.63 (0.53-0.73) |
| PF02517.13 | Abi | 47 | 13 | 34 | 0.63 (0.43-0.82) |
| PF07690.13 | MFS_1 | 378 | 45 | 333 | 0.62 (0.54-0.71) |
| PF00854.18 | PTR2 | 103 | 17 | 86 | 0.62 (0.49-0.76) |
| PF01627.20 | Hpt | 14 | 3 | 11 | 0.62 (0.30-0.94) |
| PF00361.17 | Proton_antipo_M | 648 | 19 | 629 | 0.62 (0.51-0.72) |
| PF02592.12 | Vut_1 | 34 | 7 | 27 | 0.61 (0.39-0.84) |
| PF01758.13 | SBF | 126 | 22 | 104 | 0.61 (0.48-0.74) |
| PF00860.17 | Xan_ur_permease | 559 | 115 | 444 | 0.61 (0.55-0.67) |
| PF02702.14 | KdpD | 24 | 3 | 21 | 0.61 (0.22-1.00) |
| PF13231.3 | PMT_2 | 111 | 8 | 103 | 0.61 (0.43-0.78) |
| PF00474.14 | SSF | 124 | 9 | 115 | 0.60 (0.45-0.76) |
| PF00916.17 | Sulfate_transp | 194 | 41 | 153 | 0.60 (0.50-0.70) |
| PF12821.4 | ThrE_2 | 6 | 1 | 5 | 0.60 (0.60-0.60) |
| PF03631.12 | Virul_fac_BrkB | 160 | 61 | 99 | 0.60 (0.51-0.69) |
| PF05661.9 | DUF808 | 58 | 20 | 38 | 0.60 (0.45-0.74) |
| PF03600.13 | CitMHS | 71 | 9 | 62 | 0.60 (0.37-0.82) |
| PF07786.9 | DUF1624 | 42 | 10 | 32 | 0.60 (0.39-0.80) |
| PF02447.13 | GntP_permease | 139 | 12 | 127 | 0.59 (0.44-0.75) |
| PF01914.14 | MarC | 77 | 6 | 71 | 0.59 (0.37-0.81) |
| PF01384.17 | PHO4 | 178 | 11 | 167 | 0.59 (0.39-0.79) |
| PF03594.10 | BenE | 13 | 2 | 11 | 0.59 (0.37-0.81) |
| PF03073.12 | TspO_MBR | 100 | 29 | 71 | 0.59 (0.48-0.70) |
| PF04241.12 | DUF423 | 116 | 43 | 73 | 0.59 (0.48-0.70) |
| PF09335.8 | SNARE_assoc | 429 | 51 | 378 | 0.59 (0.51-0.67) |
| PF01554.15 | MatE | 659 | 101 | 558 | 0.59 (0.53-0.64) |
| PF02233.13 | PNTB | 175 | 29 | 146 | 0.58 (0.46-0.71) |
| PF01618.13 | MotA_ExbB | 32 | 14 | 18 | 0.58 (0.36-0.81) |
| PF17113.2 | AmpE | 7 | 3 | 4 | 0.58 (0.07-1.00) |
| PF01061.21 | ABC2_membrane | 328 | 48 | 280 | 0.58 (0.51-0.65) |
| PF01292.17 | Ni_hydr_CYTB | 131 | 58 | 73 | 0.57 (0.47-0.67) |
| PF05232.9 | BTP | 8 | 1 | 7 | 0.57 (0.57-0.57) |
| PF01066.18 | CDP-OH_P_transf | 436 | 124 | 312 | 0.57 (0.51-0.63) |
| PF00672.22 | HAMP | 130 | 40 | 90 | 0.57 (0.46-0.67) |
| PF00892.17 | EamA | 813 | 125 | 688 | 0.57 (0.51-0.62) |
| PF07681.9 | DoxX | 108 | 47 | 61 | 0.56 (0.45-0.67) |
| PF03547.15 | Mem_trans | 292 | 18 | 274 | 0.56 (0.40-0.71) |
| PF01148.17 | CTP_transf_1 | 161 | 38 | 123 | 0.56 (0.45-0.66) |
| PF03006.17 | HlyIII | 93 | 7 | 86 | 0.55 (0.35-0.76) |
| PF00939.16 | Na_sulph_symp | 109 | 19 | 90 | 0.55 (0.43-0.68) |
| PF01169.16 | UPF0016 | 92 | 3 | 89 | 0.54 (0.16-0.91) |
| PF02694.12 | UPF0060 | 21 | 2 | 19 | 0.53 (0.30-0.76) |
| PF00083.21 | Sugar_tr | 285 | 46 | 239 | 0.52 (0.43-0.61) |
| PF01434.15 | Peptidase_M41 | 51 | 7 | 44 | 0.52 (0.34-0.70) |
| PF02719.12 | Polysacc_synt_2 | 62 | 19 | 43 | 0.52 (0.35-0.69) |
| PF07947.11 | YhhN | 64 | 10 | 54 | 0.52 (0.37-0.67) |
| PF01940.13 | DUF92 | 66 | 3 | 63 | 0.52 (0.26-0.77) |
| PF00893.16 | Multi_Drug_Res | 160 | 7 | 153 | 0.52 (0.35-0.68) |
| PF02628.12 | COX15-CtaA | 133 | 50 | 83 | 0.52 (0.42-0.61) |
| PF00507.16 | Oxidored_q4 | 159 | 4 | 155 | 0.51 (0.18-0.85) |
| PF06580.10 | His_kinase | 49 | 9 | 40 | 0.51 (0.28-0.74) |
| PF02040.12 | ArsB | 26 | 7 | 19 | 0.50 (0.27-0.74) |
| PF01553.18 | Acyltransferase | 20 | 4 | 16 | 0.50 (0.12-0.88) |
| PF03253.11 | UT | 9 | 2 | 7 | 0.50 (0.00-1.00) |
| PF06168.8 | DUF981 | 6 | 2 | 4 | 0.50 (0.00-1.00) |
| PF09997.6 | DUF2238 | 13 | 5 | 8 | 0.50 (0.16-0.84) |
| PF04304.10 | DUF454 | 69 | 14 | 55 | 0.49 (0.30-0.68) |
| PF06271.9 | RDD | 84 | 20 | 64 | 0.49 (0.33-0.66) |
| PF14667.3 | Polysacc_synt_C | 181 | 8 | 173 | 0.49 (0.35-0.64) |
| PF06480.12 | FtsH_ext | 42 | 6 | 36 | 0.49 (0.32-0.66) |
| PF02096.17 | 60KD_IMP | 256 | 43 | 213 | 0.49 (0.40-0.57) |
| PF03806.10 | ABG_transport | 29 | 8 | 21 | 0.49 (0.25-0.72) |
| PF07664.9 | FeoB_C | 103 | 15 | 88 | 0.49 (0.28-0.69) |
| PF09515.7 | Thia_YuaJ | 16 | 6 | 10 | 0.48 (0.12-0.85) |
| PF01595.17 | DUF21 | 238 | 50 | 188 | 0.48 (0.39-0.57) |
| PF01943.14 | Polysacc_synt | 189 | 8 | 181 | 0.47 (0.28-0.66) |
| PF02133.12 | Transp_cyt_pur | 21 | 4 | 17 | 0.46 (0.11-0.80) |
| PF03606.12 | DcuC | 37 | 4 | 33 | 0.45 (0.05-0.86) |
| PF01773.17 | Nucleos_tra2_N | 110 | 18 | 92 | 0.44 (0.28-0.61) |
| PF07662.10 | Nucleos_tra2_C | 110 | 18 | 92 | 0.44 (0.28-0.61) |
| PF13807.3 | GNVR | 25 | 3 | 22 | 0.44 (0.16-0.71) |
| PF01062.18 | Bestrophin | 10 | 2 | 8 | 0.44 (0.00-0.90) |
| PF02163.19 | Peptidase_M50 | 43 | 4 | 39 | 0.44 (0.12-0.75) |
| PF02706.12 | Wzz | 34 | 4 | 30 | 0.43 (0.20-0.67) |
| PF01027.17 | Bax1-I | 68 | 16 | 52 | 0.42 (0.26-0.58) |
| PF04235.9 | DUF418 | 7 | 1 | 6 | 0.42 (0.42-0.42) |
| PF02397.13 | Bac_transf | 11 | 1 | 10 | 0.40 (0.40-0.40) |
| PF00953.18 | Glycos_transf_4 | 86 | 3 | 83 | 0.39 (0.18-0.61) |
| PF01790.15 | LGT | 75 | 12 | 63 | 0.39 (0.21-0.58) |
| PF01252.15 | Peptidase_A8 | 63 | 22 | 41 | 0.39 (0.23-0.55) |
| PF10604.6 | Polyketide_cyc2 | 12 | 5 | 7 | 0.39 (0.11-0.66) |
| PF07264.8 | EI24 | 73 | 38 | 35 | 0.36 (0.23-0.50) |
| PF04138.11 | GtrA | 18 | 2 | 16 | 0.36 (0.00-0.79) |
| PF17154.1 | GAPES3 | 8 | 1 | 7 | 0.36 (0.36-0.36) |
| PF07298.8 | NnrU | 43 | 5 | 38 | 0.35 (0.15-0.56) |
| PF16401.2 | DUF5009 | 12 | 3 | 9 | 0.35 (0.04-0.66) |
| PF03609.11 | EII-Sor | 22 | 12 | 10 | 0.33 (0.10-0.57) |
| PF12811.4 | BaxI_1 | 20 | 2 | 18 | 0.33 (0.02-0.65) |
| PF04116.10 | FA_hydroxylase | 85 | 7 | 78 | 0.31 (0.04-0.59) |
| PF02417.12 | Chromate_transp | 8 | 1 | 7 | 0.29 (0.29-0.29) |
| PF01059.14 | Oxidored_q5_N | 27 | 2 | 25 | 0.20 (0.00-0.50) |
| PF06942.9 | GlpM | 6 | 5 | 1 | 0.20 (0.20-0.20) |
| PF07331.8 | TctB | 6 | 1 | 5 | 0.20 (0.20-0.20) |
| PF06800.9 | Sugar_transport | 29 | 2 | 27 | 0.19 (0.05-0.33) |
| PF02654.12 | CobS | 6 | 1 | 5 | 0.00 (0.00-0.00) |
| PF06966.9 | DUF1295 | 10 | 1 | 9 | 0.00 (0.00-0.00) |
1 A statistical model for improved membrane protein expression using sequence-derived features
Adapted from Saladi SM, Javed N, Müller A, Clemons WM. 2018. A statistical model for improved membrane protein expression using sequence-derived features. J Biol Chem 293:4913–4927. doi:10.1074/jbc.RA117.001052
1.1 Abstract
The heterologous expression of integral membrane proteins (IMPs) remains a major bottleneck in the characterization of this important protein class. IMP expression levels are currently unpredictable, which renders the pursuit of IMPs for structural and biophysical characterization challenging and inefficient. Experimental evidence demonstrates that changes within the nucleotide or amino-acid sequence for a given IMP can dramatically affect expression levels; yet these observations have not resulted in generalizable approaches to improve expression levels. Here, we develop a data-driven statistical predictor named IMProve, that, using only sequence information, increases the likelihood of selecting an IMP that expresses in E. coli. The IMProve model, trained on experimental data, combines a set of sequence-derived features resulting in an IMProve score, where higher values have a higher probability of success. The model is rigorously validated against a variety of independent datasets that contain a wide range of experimental outcomes from various IMP expression trials. The results demonstrate that use of the model can more than double the number of successfully expressed targets at any experimental scale. IMProve can immediately be used to identify favorable targets for characterization. Most notably, IMProve demonstrates for the first time that IMP expression levels can be predicted directly from sequence.
1.2 Introduction
The biological importance of integral membrane proteins (IMPs) motivates structural and biophysical studies that require large amounts of purified protein at considerable cost. Only a small percentage can be produced at high-levels resulting in IMP structural characterization lagging far behind that of soluble proteins; IMPs currently constitute less than 2% of deposited atomic-level structures (Hendrickson, 2016). To increase the pace of structure determination, the scientific community created large government-funded structural genomics consortia facilities, like the NIH-funded New York Consortium on Membrane Protein Structure (NYCOMPS) (Punta et al., 2009). For this representative example, more than 8000 genes, chosen based on characteristics hypothetically related to success, yielded only 600 (7.1%) highly expressing proteins (Love et al., 2010) resulting to date in 34 (5.6% of expressed proteins) unique structures (based on annotation in the RCSB PDB (Berman et al., 2000)). This example highlights the funnel problem of structural biology, where each stage of the structure pipeline eliminates a large percentage of targets compounding into an overall low rate of success (Lewinson et al., 2008). With new and rapidly advancing technologies like cryo-electron microscopy, serial femtosecond crystallography, and micro-electron diffraction, we expect that the latter half of the funnel, structure determination, will increase in success rate (Johansson et al., 2017; Merk et al., 2016; Nannenga and Gonen, 2016). However, IMP expression levels will continue to limit targets accessible for study (Bill et al., 2011).
Tools are needed for improving the number of IMPs with successful heterologous expression (we use expression synonymously for the term ‘overexpression’ in the field). While significant work has shown promise on a case-by-case basis, e.g., growth at lower temperatures, codon optimization (Nørholm et al., 2012), 5’ UTR randomization (Mirzadeh et al., 2015), and regulating transcription (Wagner et al., 2008), a generalizable solution remains elusive. Several RNA structure prediction methods have been used to explain or predict soluble protein expression levels (Price et al., 2009; Reis and Salis, 2020). For individual IMPs, simple changes, can have dramatic effects on the amount of expressed proteins—only partially explained by computational methods in the case of modifications at the 5’ region (Mirzadeh et al., 2015; Sarkar et al., 2008; Schlinkmann et al., 2012). Broadly, each new IMP target must be addressed individually as the conditions that were successful for a previous target seldom carry over to other proteins, even amongst closely related homologs (Lewinson et al., 2008; Marshall et al., 2016).
Considering the scientific value of IMP studies, it is surprising that there are no methods that can provide solutions for improved expression outcomes with broad applicability across protein families and genomes. There are no approaches that can decode sequence-level information for predicting IMP expression; yet it is common knowledge that sequence changes which alter overall biophysical features of the protein and mRNA transcript can measurably influence IMP biogenesis. While physics-based approaches which have proven successful in correlating integration efficiency and expression (Marshall et al., 2016; Niesen et al., 2017), that and other work revealed that simple application of specific ‘sequence features’ are inadequate to predict IMP expression (Daley et al., 2005; Nørholm et al., 2013). As an example, while the positive-inside rule is an important indicator of proper IMP biogenesis, the number of positive-charges on cytoplasmic loops alone does not predict expression level (Seppälä et al., 2010; Van Lehn et al., 2015). The reasons for this failure to connect sequence to expression likely lie in the complex underpinnings of IMP biogenesis, where the interplay between many sequence features at both the protein and nucleotide levels must be considered. Optimizing for a single sequence feature likely diminishes the beneficial effect of other features (e.g., increasing TM hydrophobicity might diminish favorable mRNA properties). Without accounting for the broad set of sequence features related to IMP expression, it is impossible to predict differences in expression levels.
Development of a low-cost, computational resource that significantly and reliably predicts improved expression outcomes would transform the study of IMPs. Attempts to develop such algorithms have so far failed. Several examples, Daley, von Heijne, and coworkers (Daley et al., 2005; Nørholm et al., 2013) as well as NYCOMPS, were unable to use experimental expression data sets to train models that returned any predictive performance (personal communication). This is not surprising, given the difficulty of expressing IMPs and the limits in the knowledge of the sequence features that drive expression. In other contexts, statistical tools based on sequence have been shown to work; for example, those developed to predict soluble protein expression and/or crystallization propensities (Bertone et al., 2001; Jahandideh et al., 2014; Price et al., 2009). Such predictors are primarily based on available experimental results from the Protein Structure Initiative (Chen et al., 2004; Gabanyi et al., 2011). While collectively these methods have supported significant advances in biochemistry, none of the models are able to predict IMP outcomes due to limitations inherent in the model development process. As IMPs have an extremely low success rate, they are either explicitly excluded from the training process or are implicitly down-weighted by the statistical model (for representative methodology see (Slabinski et al., 2007a)). Consequently, none have successfully been able to map IMP expression to sequence.
Here, we demonstrate for the first time that it is possible to predict IMP expression directly from sequence. The resulting predictor allows one to enrich expression trials for proteins with a higher probability of success. To connect sequence to prediction, we develop a statistical model that maps a set of sequences to experimental expression levels via calculated features—thereby simultaneously accounting for the many potential determinants of expression. The resulting IMProve model allows ranking of any arbitrary set of IMP sequences in order of their relative likelihood of successful expression. The IMProve model is extensively validated against a variety of independent datasets demonstrating that it can be used broadly to predict the likelihood of expression in E. coli of any IMP. With IMProve, we have built a way for more than two-fold enrichment of positive expression outcomes relative to the rate attained from the current method of randomly selecting targets. We highlight how the model informs on the biological underpinnings that drive likely expression. Finally, we provide direct examples where the model can be used for a typical researcher. Our novel approach and the resulting IMProve model provide an exciting paradigm for connecting sequence space to complex experimental outcomes.
1.3 Results
For this study, we focus on heterologous expression in E. coli, due to its ubiquitous use as a host across the spectrum of the membrane proteome. Low cost and low barriers for adoption highlight the utility of E. coli as a broad tool if the expression problem can be overcome. Furthermore, proof-of-principle in E. coli will illustrate methodology for constructing similar predictive methods for expressing IMPs in other hosts (e.g., yeast, insect cells).
1.3.1 Development of a computational model trained on E. coli expression data
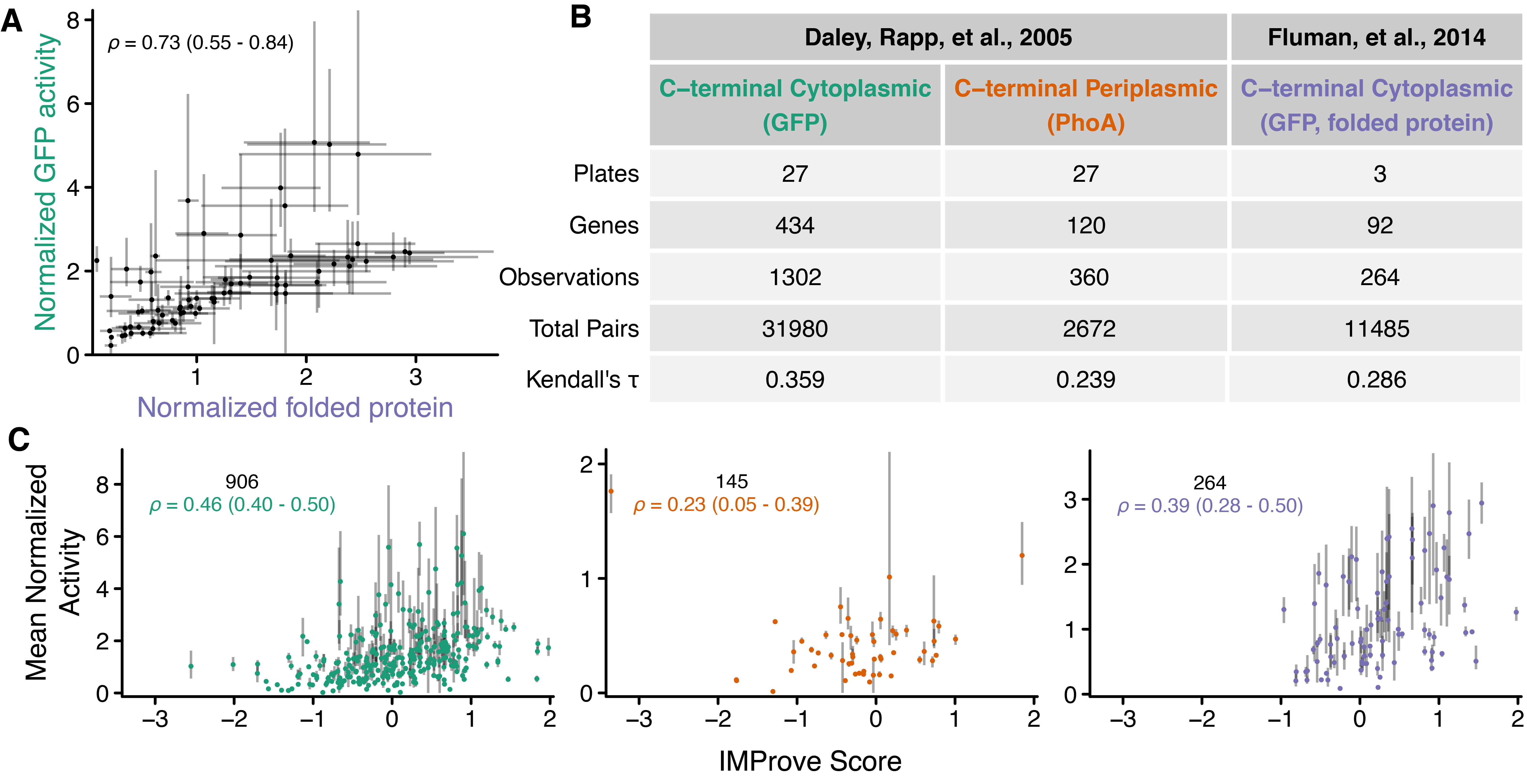
A key component of any data-driven statistical model is the choice of dataset used for training. Having searched the literature, we identified two publications that contained quantitative datasets on the IPTG-induced overexpression of E. coli polytopic IMPs in E. coli. The first set, Daley, Rapp et al., contained activity measures, proxies for expression level, from C-terminal tags of either GFP or PhoA (alkaline phosphatase)(Daley et al., 2005). The second set, Fluman et al., used a subset of constructs from the first and contained a more detailed analysis utilizing in-gel fluorescence to measure folded protein (Fluman et al., 2014) (see Section 1.5.4). The expression levels strongly correlated (Spearman’s \(\rho\) = 0.73) between the two datasets demonstrating that normalized GFP activity was a good measure of the amount of folded IMP (Figure 1.1A and (Fluman et al., 2014; Geertsma et al., 2008)). The experimental set-up employed multiple 96-well plates over multiple days resulting in pronounced variability in the absolute expression level of a given protein between trials. Daley, Rapp et al. calculated average expression levels by dividing the raw expression level of each protein by that of a control protein on the corresponding plate.
To successfully map sequence to expression, we additionally needed to derive numerical features from a given gene sequence that have been empirically related to expression. 87 sequence features from protein and nucleotide sequence were calculated for each gene using custom code together with published software (codonW (Peden, 2000), tAI (dos Reis et al., 2003), NUPACK (Zadeh et al., 2011), Vienna RNA (Lorenz et al., 2011), Codon Pair Bias (Coleman et al., 2008), Disembl (Linding et al., 2003), and RONN (Yang et al., 2005)). Relative metrics (e.g., codon adaptation index) are calculated with respect to the E. coli K-12 substr. MG1655 (Zhou and Rudd, 2013) quantity. The octanol-water partitioning (Wimley et al., 1996), GES hydrophobicity (Engelman et al., 1986), \(\Delta G\) of insertion (Hessa et al., 2007) scales were employed as well. Transmembrane segment topology was predicted using Phobius constrained for the training data and Phobius for all other datasets (Käll et al., 2004). Two RNA secondary structure metrics were prompted in part by Goodman, et al. (Goodman et al., 2013). Table 1.1 includes a detailed description of each feature. All features are calculated solely from the coding region of each gene of interest excluding other portions of the open reading frame and plasmid (e.g., linkers and tags, 5’ untranslated region, copy number).
Fitting the data to a simple linear regression provides a facile method for deriving a weight for each feature. However, using the set of sequence features, we were unable to successfully fit a linear regression using the normalized GFP and PhoA measurements reported in the Daley, Rapp et al. study. Similarly, using the same feature set and data, we were unable to train a standard linear Support Vector Machine (SVM) to predict the expression data either averaged or across all plates (see Table 1.1; Section 1.5.2, Section 1.5.3). Due to the attempts by others to fit this data, this outcome may not be surprising and suggested that a more complex analysis was required.
We hypothesized that training on relative measurements across the entire dataset introduced errors that were limiting. To address this, we instead only compare measurements within an individual plate, where differences between trials are less likely to introduce errors. To account for this, a preference-ranking linear SVM algorithm (SVMrank (Tsochantaridis et al., 2005)) was chosen (see Section 1.5.4). Simply put, the SVMrank algorithm determines the optimal weight for each sequence feature to best rank the order of expression outcomes within each plate over all plates, which results in a model where higher expressing proteins have higher scores. The outcome is identical in structure to a multiple linear regression, but instead of minimizing the sum of squared residuals, the SVMrank cost function accounts for the plate-wise constraint specified above. In practice, the process optimizes the correlation coefficient Kendall’s \(\tau\) (Equation 1.1) to converge upon a set of weights.
\[ \tau_{\text{kendall}} = \frac{\text{N}_\text{correctly ordered pairs} - \text{N}_\text{swapped pairs}}{\text{N}_\text{total pairs}} \tag{1.1}\]
The SVMrank training metric shows varying agreement for all three groups (i.e., \(\tau_{\text{kendall}} > 0\)) (Figure 1.1B). For individual genes, activity values normalized and averaged across trials were not directly used for the training procedure (see Section 1.5.4); yet one would anticipate that scores for each gene should broadly correlate with the expression average. Indeed, the observed normalized activities positively correlate with the score (dubbed IMProve score for Integral Membrane Protein expression improvement) output by the model (Figure 1.1C, \(\rho > 0\)). Since SVMrank transforms raw expression levels within each plate to ranks before training, Spearman’s \(\rho\), a rank correlation coefficient describing the agreement between two ranked quantities, is better suited for quantifying correlation over more common metrics like the \(R^2\) of a regression and Pearson’s \(r\). While these metrics demonstrate that the model can rank expression outcomes across all proteins in the training set, for PhoA-tagged proteins, the model appears less successful. The implications for this are discussed later (see Figure 1.2G).
1.3.2 Demonstration of prediction against an independent large expression dataset
While the above analyses show that the model successfully fits the training data, we assess the broader applicability of the model outside the training set based on its success at predicting the outcomes of independent expression trials from distinct groups and across varying scales. The first test considers results from NYCOMPS, where 8444 IMP genes entered expression trials, in up to eight conditions, resulting in 17114 expression outcomes (Figure 1.2A) (Punta et al., 2009). The majority of genes were attempted in only one condition (Figure 1.2B), and, importantly, outcomes were non-quantitative (binary: expressed or not expressed) as indicated by the presence of a band by Coomassie staining of an SDS-PAGE gel after small-scale expression, solubilization, and nickel affinity purification (Love et al., 2010). For this analysis, the experimental results are either summarized as outcomes per gene or broken down as raw outcomes across defined expression conditions. For outcomes per gene, we can consider various thresholds for considering a gene as positive based on NYCOMPS expression success (Figure 1.2B). The most stringent threshold only regards a gene as positive if it has no negative outcomes (“Only Positive”, Figure 1.2B, red). Since a well expressing gene would generally advance in the NYCOMPS pipeline without further small-scale expression trials, this positive group likely contains the best expressing proteins. A second category comprises genes with at least one positive and at least one negative trial (“Mixed”, Figure 1.2B, blue). These genes likely include proteins that are more difficult to express.
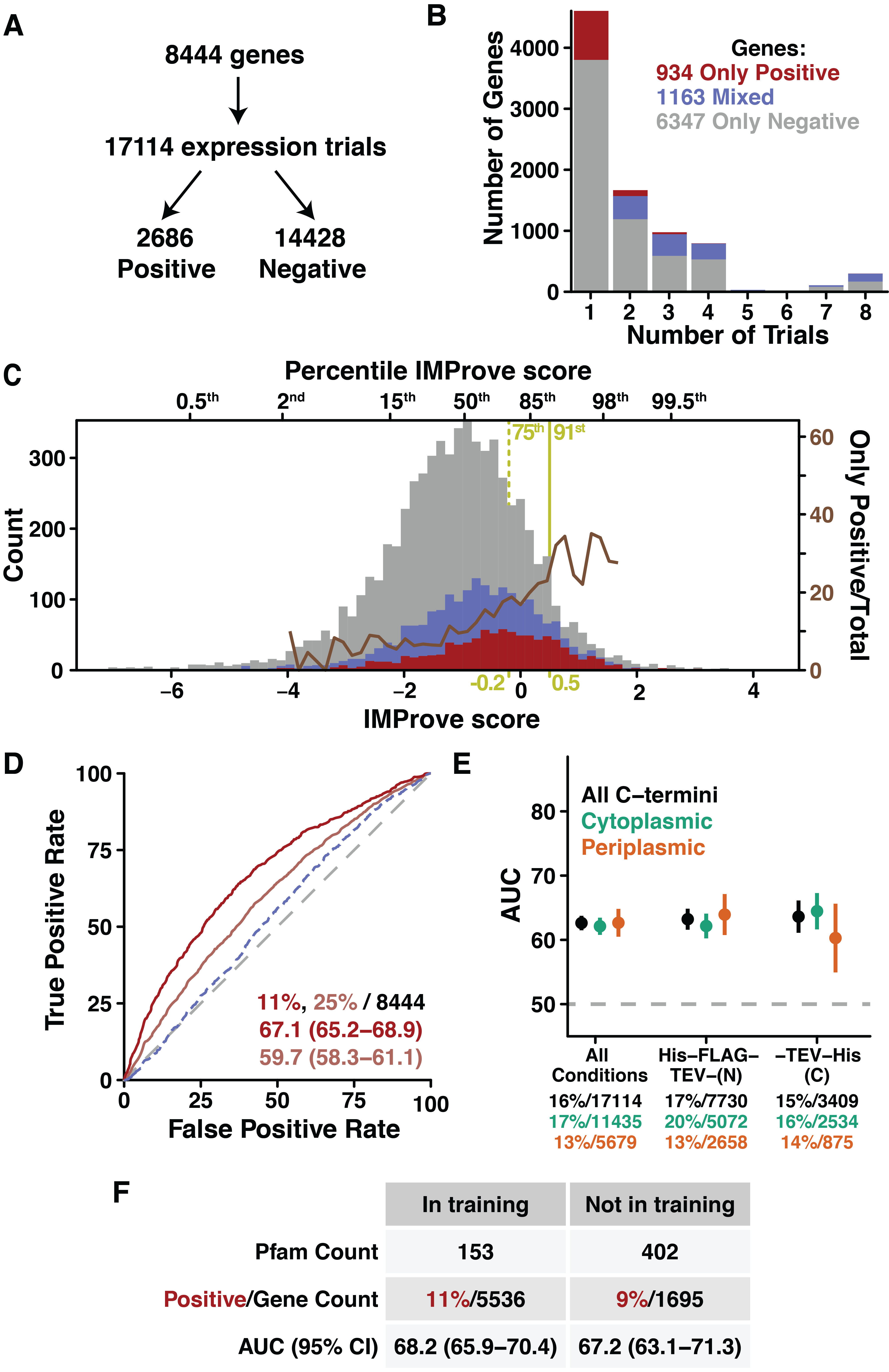
A histogram of the IMProve scores for genes separated by expression success shows predictive power of the IMProve model (Only Positive, red; Mixed, blue; Only Negative, grey) (Figure 1.2C). Visually, the distribution of the scores for the Only Positive group is shifted to a higher score relative to the Only Negative and Mixed groups. The dramatic increase in the percentage of Only Positive genes as a function of increasing IMProve score (overlaid as a brown line) further emphasizes this. In fact, simply selecting the top 25% of proteins by IMProve score (-0.2 or higher, Figure 1.2C, yellow dotted line) would have increased the number of positive outcomes from 11% to 20%, a 1.8 fold improvement.
While this is suggestive, we aim to more systematically assess predictive power. For NYCOMPS and subsequent data sets, the outcomes are either binary (e.g., in NYCOMPS expressed or not expressed) or categorical (e.g., arbitrary ranked scale), correlation coefficients cannot be used here. Instead, we turn to an alternative framework: the Receiver Operating Characteristic (ROC). ROC curves quantify the tradeoff between true positive and false positive predictions across numerical scores from a predictor. The figure of merit is the Area Under the ROC Curve (AUC), where a perfect predictor has an AUC = 100% and a random predictor has an AUC = 50 % (Figure 1.2D, grey dashed line). An AUC greater than 50% indicates that a model is predictive (percentage signs hereafter omitted) (see Section 1.5.5 and (Swets et al., 2000)). We will use the ROC framework to quantitatively validate the ability of our model to predict protein expression data—all independent of the training process.
Returning to the NYCOMPS data, ROC analysis shows that the IMProve model markedly distinguishes genes in the most stringent positive group (Only Positive) from all other genes (AUC = 67.1) (Figure 1.2C red). A permissive threshold considering genes as positive with at least one positive trial (Only Positive plus Mixed genes) shows moderate predictive power (Figure 1.2C pink, AUC = 59.7). If instead the Mixed genes are considered alone (excluding the Only Positive), the model very weakly distinguishes the Mixed group from the Only Negative genes (Figure 1.2C dashed blue, AUC = 53.5). This likely supports the notion that the Mixed pool largely consists of more difficult-to-express genes.
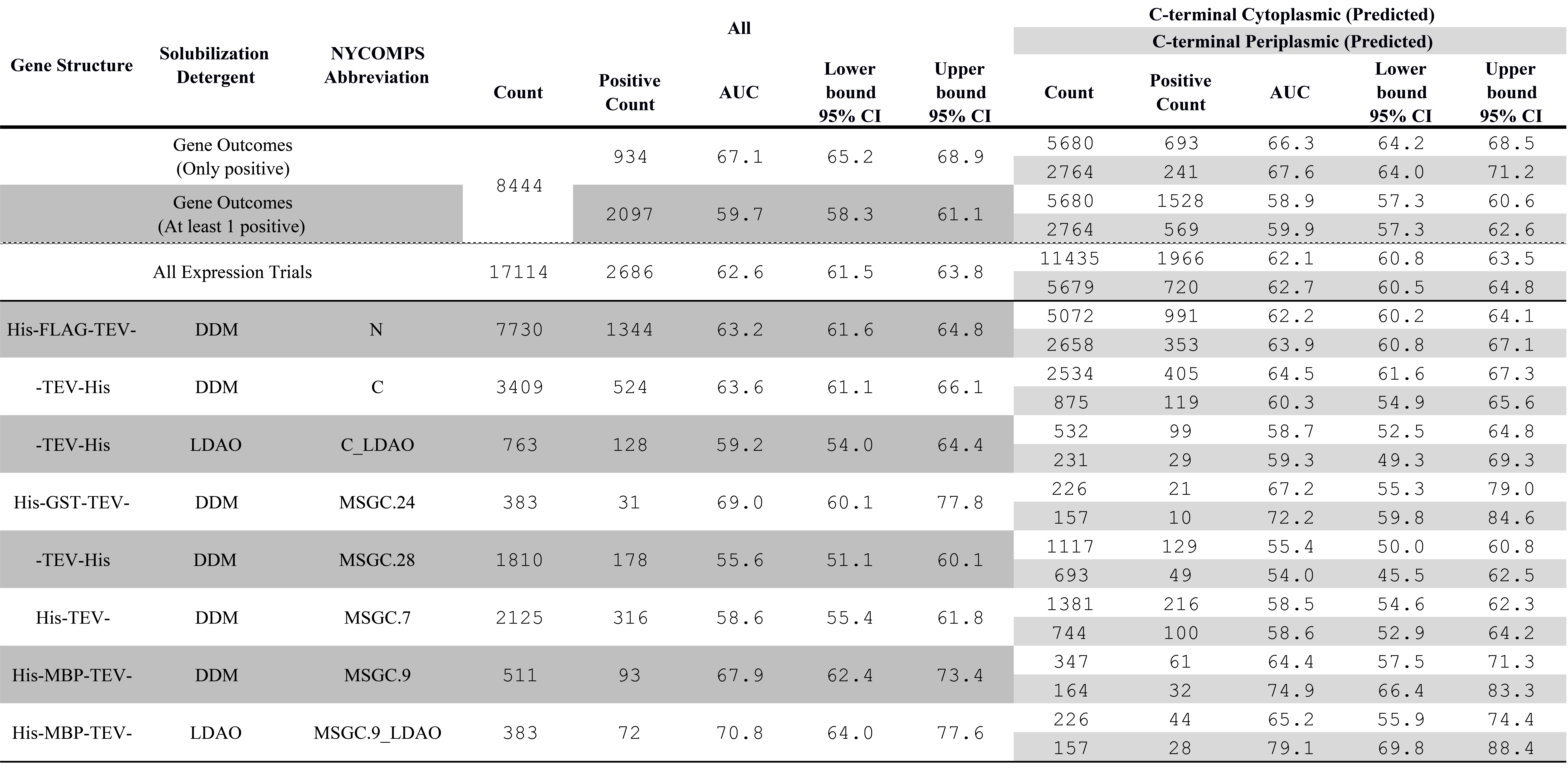
We next confirm the ability of the IMProve model to predict within plasmids or sequence space distinct from those within the limited training set. For an overfit model, one might expect that only the subset of targets which most closely mirror the training data would show strong prediction. On the contrary, the model shows consistent performance throughout each of the eight distinct experimental conditions tested (Figure 1.2G and Table 1.2). One may also consider that the small size of the training set limited the number of protein folds sampled and, therefore, limited the number of folds that could be predicted by the model. To test this, we consider the performance of the model with regards to protein homology families, as defined by Pfam family classifications (Finn et al., 2014). The 8444 genes in the NYCOMPS dataset fall into 555 Pfam families (~15% not classified). To understand whether the IMProve score is biased towards families present in the training set, we separate genes in the NYCOMPS dataset into either within the 153 Pfam families found in the training set or outside this pool (i.e., not in the training set). Satisfyingly, there is no significant difference in AUC at 95% confidence between these groups (68.2 versus 67.2) (Figure 1.2H). Combined, this highlights that the model is not sensitive to the experimental design of the training set and predicts broadly across different vector backbones and protein folds.
The ability to predict the experimental data from NYCOMPS allows returning to the question of alkaline phosphatase as a metric for expression. For the training set, proteins with C-termini in the periplasm show weaker fitting by the model (Figure 1.1, orange). To assess the generality of this result, the NYCOMPS outcomes are split into pools for either cytoplasmic or periplasmic C-terminal localization and AUCs are calculated for each. There are no significant differences in predictive capacity across all conditions (Figure 1.2G, green vs. orange) irrespective of whether the tag is at the N- or C-terminus. This demonstrates that the IMProve model is applicable for all topologies.
At this point, it is useful to consider the potential improvement in the number of positive outcomes by using the IMProve model. NYCOMPS tested about a tenth of the 74 thousand possible IMPs from the 98 genomes of interest for expression (Punta et al., 2009). Had NYCOMPS tested the same number of genes from this pool, but selected to have an IMProve score greater than 0.5 (at the 91st percentile (Figure 1.2C, yellow solid line)), they would have increased their positive pool of 934 by an additional 1207 proteins. This represents a more than two-fold improvement in the return on investment and is a clear benchmark of success for the IMProve model.
1.3.3 Further demonstration of prediction against small-scale independent datasets
The NYCOMPS example demonstrates the predictive power of the model across the broad range of sequence space encompassed by that dataset. Next, the performance of the model is tested against relevant subsets of sequence space (e.g., a family of proteins or the proteome from a single organism), which are reminiscent of laboratory-scale experiments that precede structural or biochemical analyses. While a number of datasets exist (Bernaudat et al., 2011; Eshaghi et al., 2005; Gordon et al., 2008; Korepanova et al., 2005; Lewinson et al., 2008; Lundstrom et al., 2006; Ma et al., 2013; Madhavan et al., 2010; Petrovskaya et al., 2010; Surade et al., 2006; Szakonyi et al., 2007), we identified seven for which complete sequence information could be obtained to calculate all the necessary sequence features (Korepanova et al., 2005; Lundstrom et al., 2006; Ma et al., 2013; Madhavan et al., 2010; Surade et al., 2006).
To understand the predictive performance within each of the small-scale datasets, we analyze the predictive performance of the model and highlight how the model could have been used to streamline those experiments. The clear predictive performance within the large-scale NYCOMPS dataset (Figure 1.2) serves as a benchmark of expected performance at the scale of the experimental efforts for an individual lab (Figure 1.3A). As targets within the various datasets were tested only one or a few times, experimental variability within each set could play a large-role on the outcomes noted. Therefore, we summarize positives within each dataset as those genes with the highest level of outcome as reported by the original authors as this outcome is likely most robust to such variability (e.g., seen via Coomassie Blue staining of an SDS-PAGE gel). To be complete, we have plotted and considered predictive performance across all possible outcomes as well (Figure 1.3B-D, Figure 1.6, Figure 1.7).
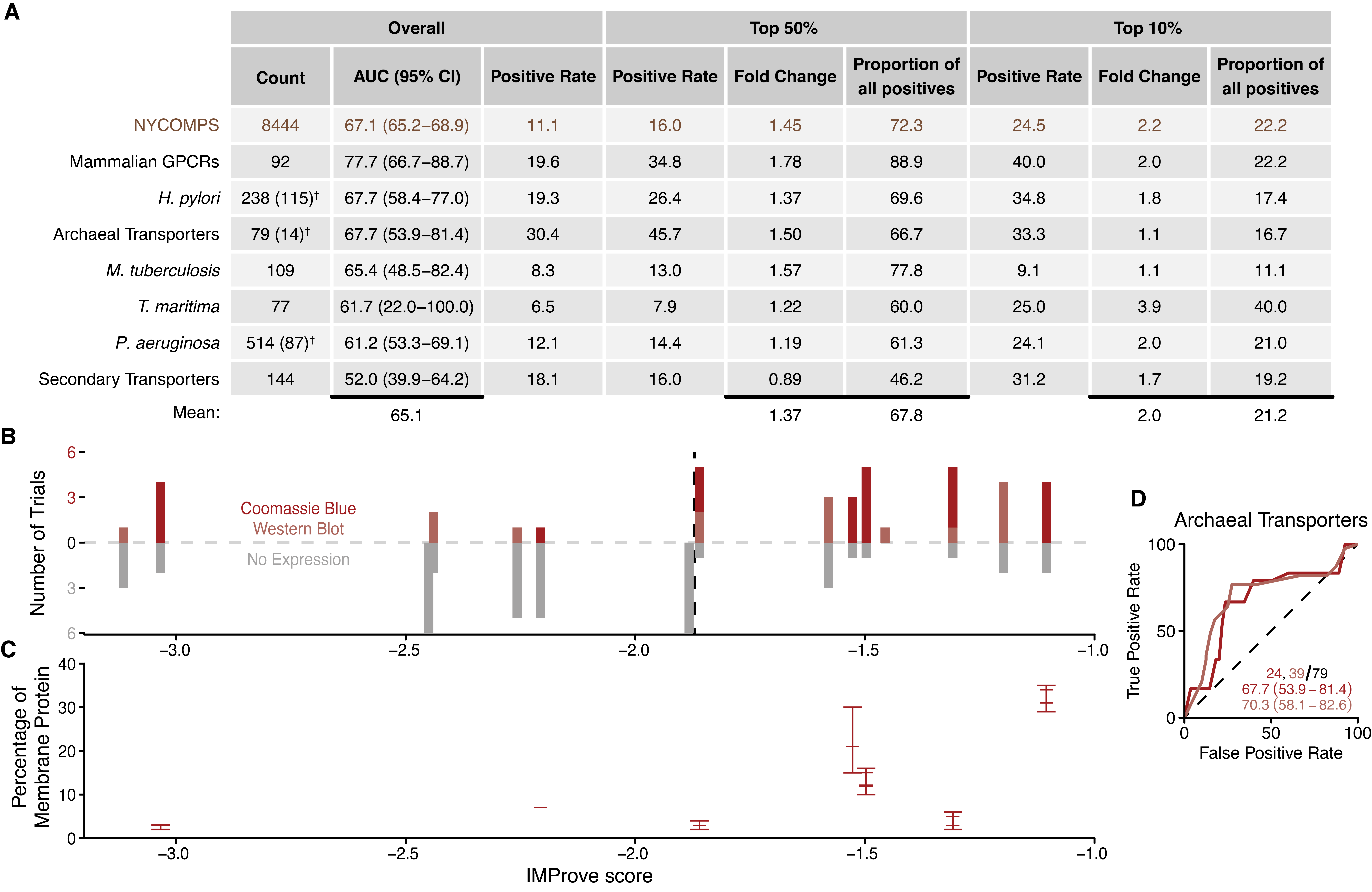
The performance of the IMProve model for each of the small-scale datasets is consistent with that seen for the NYCOMPS dataset (Figure 1.3A). This is most directly indicated by a mean AUC across all datasets of 65.6, highlighting the success across the varying scales. While the overall positive rate is different for each dataset, considering a cut-off in IMProve score, e.g., the top 50% or 10% of targets ranked by score, would have resulted in a greater percentage of positive outcomes. On average, ~70% of positives are captured within the top half of scores. Similarly, for the top 10% of scores, on average over 20% of the positives are captured. Simply put, for one tenth of the work one would capture a significant number of the positive outcomes within the pool of targets in each dataset.
For broader consideration, one can consider the fold change in positive rate by selecting targets informed by IMProve scores. Using the data available, only testing proteins within the top 10% of scores would result in an average fold change of 2.0 in the positive rate (i.e., twice as many positively expressed proteins). As positive rate is a bounded quantity (maximum is 100%), the possible fold change is bounded as well and becomes relative to the overall positive rate when considering various cut-offs (e.g., for T. maritima the maximum fold-change is 15.4 while for archaeal transporters it is 3.3). Taking the average maximum possible fold change (7.5), the IMProve model achieves nearly a third of the possible improvement in positive rate compared to a perfect predictor.
Since the IMProve model was trained on quantitative expression levels, we also expect that it captures quantitative trends in expression, i.e., a higher score translates to greater amount of expressed protein. While the NYCOMPS results are consistent with this (Figure 1.2b), of the various data sets, only the expression of archaeal transporters presents quantitative outcomes for consideration. For this dataset, 14 archaeal transporters were chosen based on their homology to human proteins (Ma et al., 2013) and tested for expression in E. coli; total protein was quantified in the membrane fraction by Coomassie Blue staining of an SDS-PAGE gel. Here, the majority of the expressing proteins fall into the higher half of the IMProve scores, 7 out of 9 of those with multiple positive outcomes (Figure 1.3B). Strikingly, quantification of the Coomassie Blue staining highlights a clear correlation with the IMProve score where the higher expressing proteins have higher scores (Figure 1.3C).
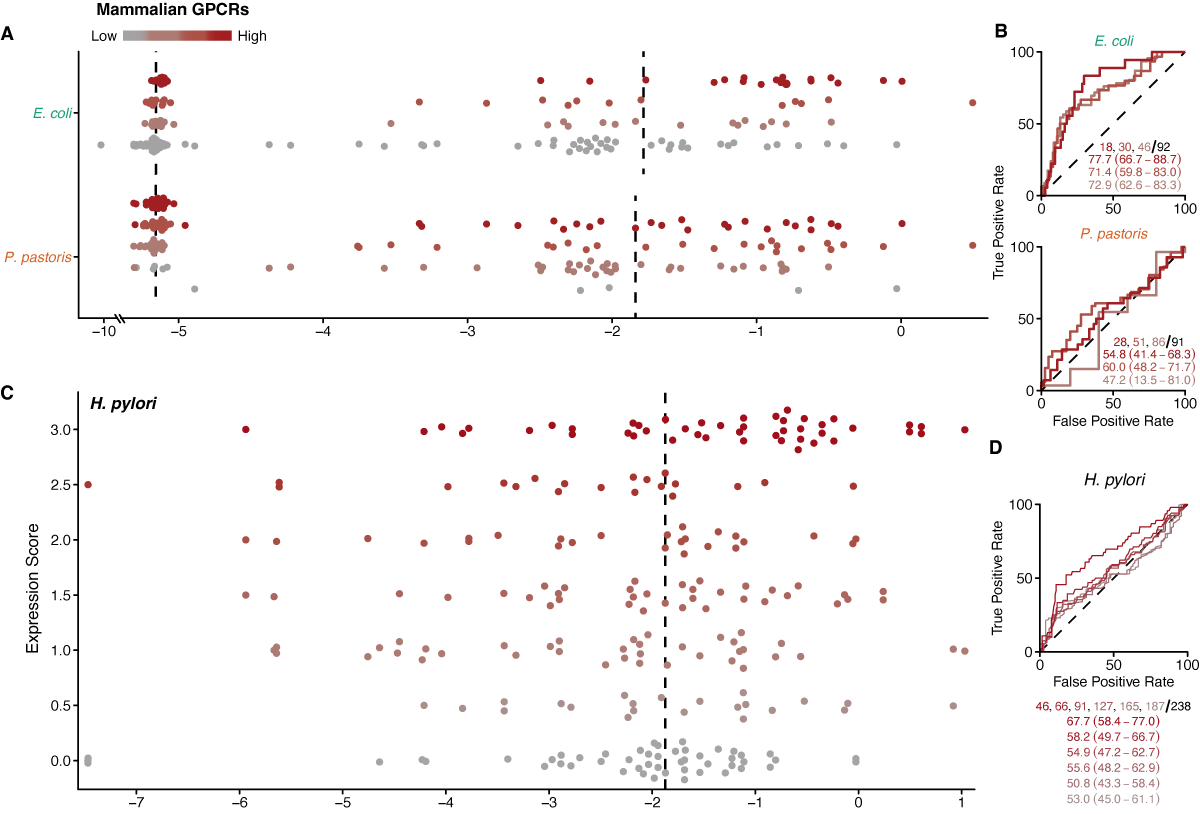
A final test considers the ability of the model to predict expression in hosts other than E. coli. The expression trials of 101 mammalian GPCRs in bacterial and eukaryotic systems (Lundstrom et al., 2006) provides a data set for considering this question. For this experiment, trials in E. coli clearly follow the trend that IMProve can predict within this group of mammalian proteins (AUC = 77.7) (Figure 1.3A, Figure 1.6A,B). However, the expression of the same set of proteins in P. pastoris fails to show any predictive performance (AUC = 54.8) (Figure 1.6A,B). This lack of predictive performance in P. pastoris suggests that the parameterization of the model, calibrated for E. coli expression, requires retraining to generate a different model that captures the distinct interplay of sequence parameters in other hosts.
1.3.4 Biological importance of various sequence features
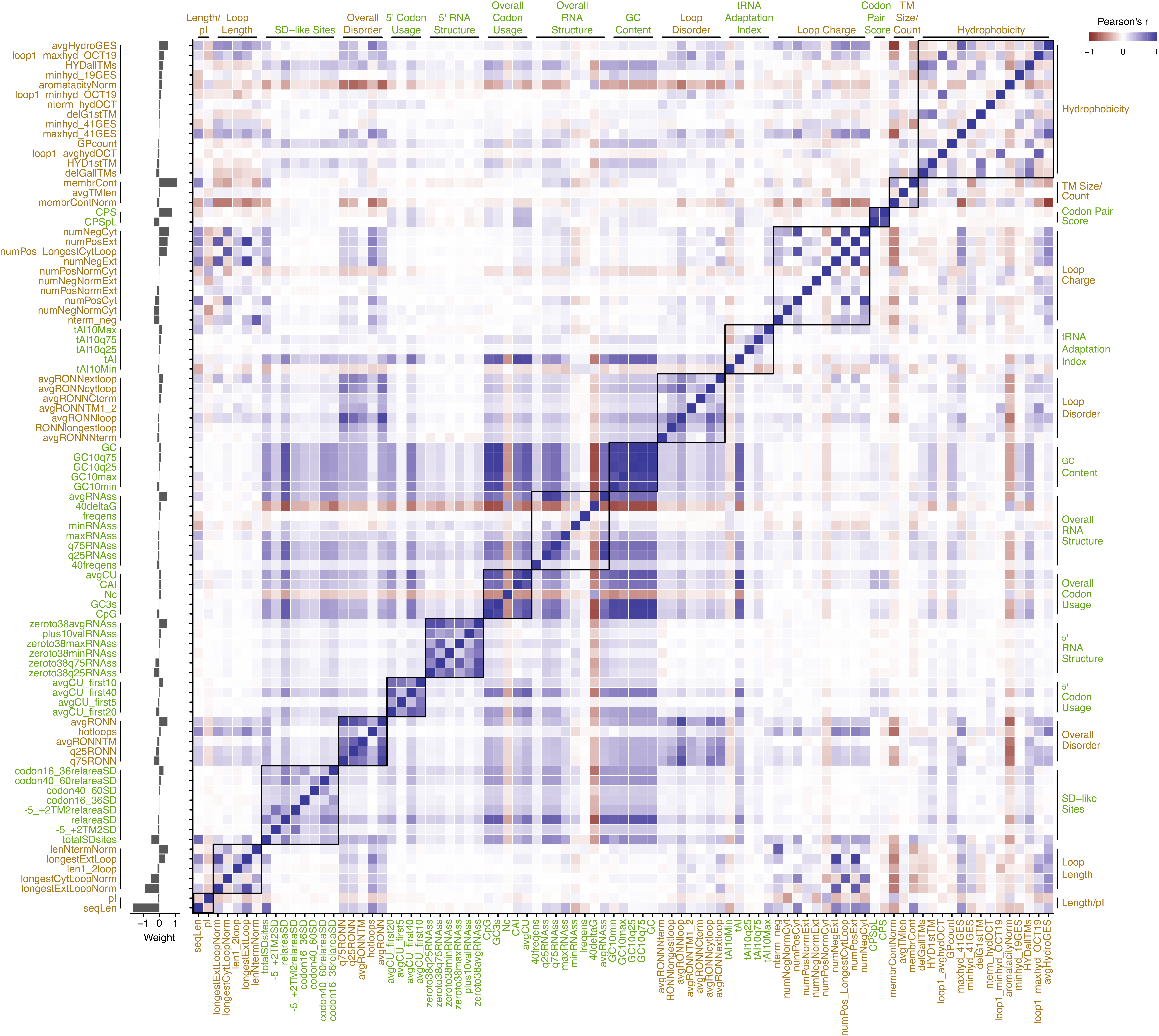
Considering the success of IMProve, one might anticipate that biological properties driving prediction may provide insight into IMP biogenesis and expression. To derive principles about which features drive prediction, most statistical models (including SVMrank as used here) require that each data point and its features are drawn identically and distributed independently from an underlying distribution (referred to as “i.i.d.”). In the case of a linear model, if the features meet the i.i.d. criteria then extracting the importance of each feature is straightforward; the weight assigned to each feature corresponds to its relative importance. However, in our case, the input features do not meet the i.i.d. criteria because significant correlation exists between individual features (Figure 1.8).
Since overlapping sets of correlating features represent biological properties, the importance of a given biological property is distributed across many features. The result is that one cannot directly determine the biological underpinnings of the IMProve score. For example, the importance of transmembrane segment hydrophobicity for membrane partitioning is likely distributed between several features: among these the average \(\Delta \text{G}_{\text{insertion}}\) (Hessa et al., 2007) of TM segments has a positive weight whereas average hydrophobicity, a correlating feature, has a negative weight (Table 1.1, Figure 1.8). While this is counterintuitive, it is consistent with the expectation that the overall importance of transmembrane segment hydrophobicity is the sum of many correlated features. While one cannot disentangle the correlation between features, we attempt to address this complication by coarsening our view of the features to two levels: First, we analyze features derived from protein versus those derived from nucleotide sequence, and then we look more closely at features groups after categorizing by biological property.
The coarsest view of the features is a comparison of those derived from protein sequence versus those derived from nucleotide sequence. The summed weight for protein features is around zero, whereas for nucleotide features the summed weight is slightly positive suggesting that in comparison these features may be more important to the predictive performance of the model (Figure 1.4A). Within the training set, protein features more completely explain the score both via correlation coefficients (Figure 1.4B). However, comparison of the predictive performance of the two subsets of weights shows that the nucleotide features alone can give similar performance to the full model for the NYCOMPS dataset (Figure 1.4C). Within the small-scale datasets investigated, using only protein or nucleotide features shows no significant difference in predictive power at 95% confidence (Figure 1.4D). In general, this suggests that neither protein nor nucleotide features are solely responsible for successful IMP expression. However, within the context of the trained model, nucleotide features are critical for predictive performance for a large and diverse dataset such as NYCOMPS. This finding corroborates growing literature that the nucleotide sequence holds significant determinants of biological processes (Chartron et al., 2016; Fluman et al., 2014; Gamble et al., 2016; Goodman et al., 2013; Li et al., 2012).
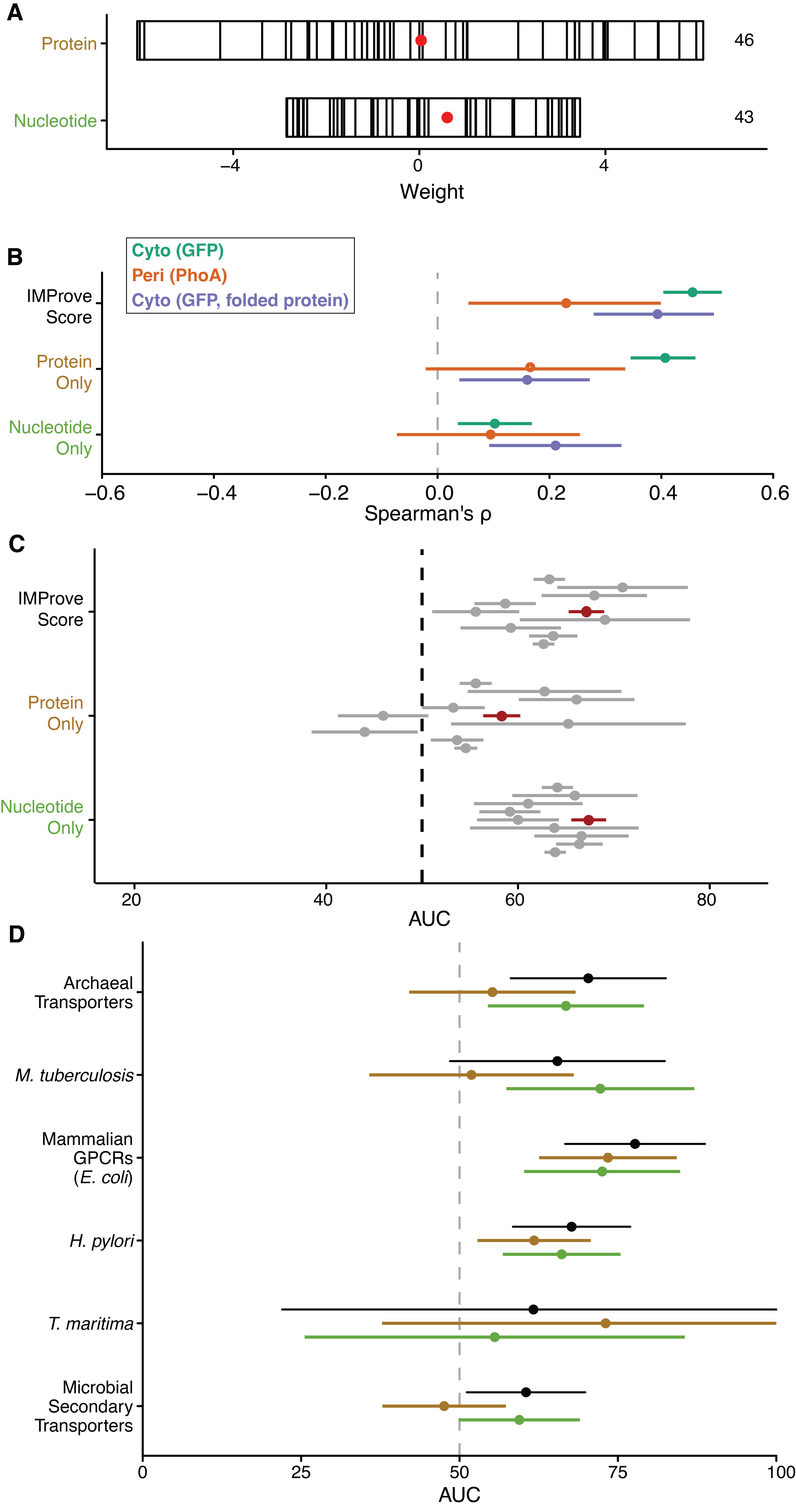
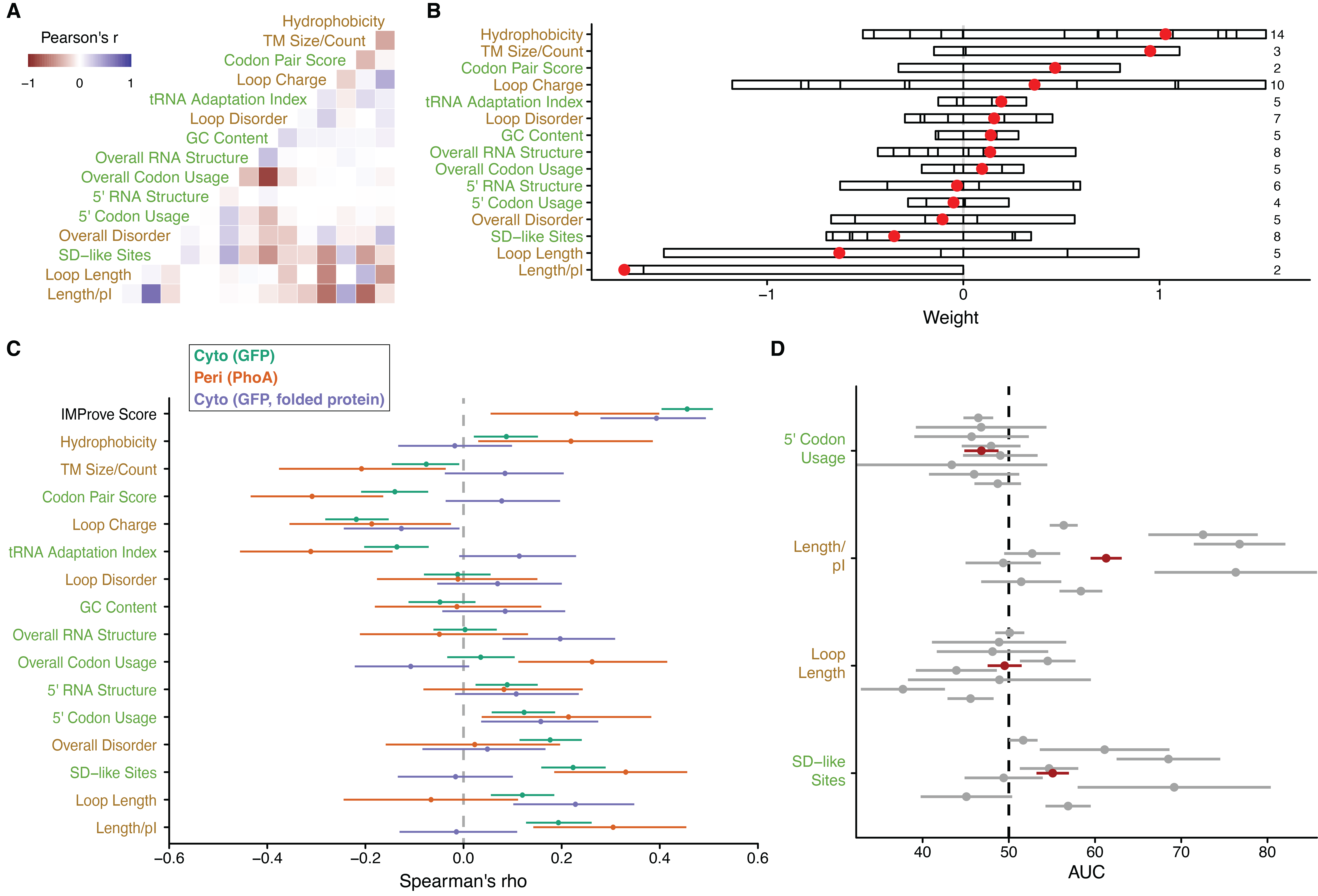
We next collapse conceptually similar features into biological categories that allow us to infer the phenomena that drive prediction. Categories are chosen empirically (e.g., the hydrophobicity group incorporates sequence features such as average hydrophobicity, maximum hydrophobicity, \(\Delta \text{G}_{\text{insertion}}\)), which results in a reduction in overall correlation (Figure 1.9A). The full category list is provided in Table 1.1. To visualize the importance of each category, the collapsed weights are summarized in Figure 1.9B, where each bar contains individual feature weights within a category. Features with a negative weight are stacked to the left of zero and those with a positive weight are stacked to the right. A red dot represents the sum of all weights, and the length of the bar gives the total absolute value of the combined weights within a category. Ranking the categories based on the sum of their weight suggests that some categories play a more prominent role than others. These include properties related to transmembrane segments (hydrophobicity and TM size/count), codon pair score, loop length, and overall length/pI.
To explore the role of each biological category in prediction, the performance of the model is assessed using only features within a given category. First, the strength of the correlation coefficients for given categories within the training set suggests the relative utility of each category for prediction. (Figure 1.9C, as in Figure 1.4B). Examples of categories with high correlation coefficients are 5’ Codon Usage, Length/pI, Loop Length, and SD-like Sites. To verify their importance for prediction, we consider the AUC for prediction using each feature category for the NYCOMPS dataset (Figure 1.9D). In this analysis, only Length/pI shows some predictive power. Overall, the analysis of the training and large-scale testing dataset shows that no feature category independently drives the predictor. Excluding each individually does not significantly affect the overall predictive performance, except for Length/pI (data not shown). Sequence length composes the majority of the weight within this category and is one of the highest weighted features in the model (Figure 1.9A). This is consistent with the anecdotal observation that larger IMPs are typically harder to express. However, this parameter alone would not be useful for predicting within a smaller subset, like a single protein family, where there is little variance in length (e.g., Figure 1.3,Figure 1.5). One might develop a predictor that was better for a given protein family under certain conditions with a subset of the entire features considered here; yet this would require a priori knowledge of the system, i.e., which sequence features were truly most important, and would preclude broad generalizability as shown for the IMProve model.
1.3.5 Usage of the IMProve model for IMP expression
We illustrate the IMProve model’s ability to identify promising homologs within a protein family by considering subsets of the broad range of targets tested by NYCOMPS. First, we consider two examples for protein families that do not have associated atomic resolution structures: copper resistance proteins (CopD, PF05425) and short-chain fatty-acid transporters (AtoE, PF02667). In the first two rows of Figure 1.5A, genes from the two families are plotted by IMProve score and colored by experimental outcome. In both cases, as indicated by the AUCs of 88.2 and 80.7 (Figure 1.5A), the model excels at predicting these families and provides a clear score cut-off to guide target selection for future expression experiments. For example, we expect that CopD homologs with IMProve scores above -1 will have a higher likelihood of expressing over other homologs.

We have calculated predictive performance for each Pfam found in the NYCOMPS data which allows us to provide considerations for future experiments (Table 1.3). In particular, we highlight three families with many genes tested, multiple experimental trials and a spread of outcomes: voltage-dependent anion channels (PF03595), Na/H exchangers (PF00999), and glycosyltransferases (PF00535). For these, a very clear IMProve score cut-off emerges from the experimental outcomes (dashed line in Figure 1.5A). Strikingly, for these families the IMProve model clearly ranks the targets with Only Positive outcomes (red) at higher scores, again suggesting a preference for the best expressing proteins (see Figure 1.2 and Figure 1.3). Similarly, many more families within NYCOMPS are predicted with high statistical confidence (Table 1.3); we provide a subset as Figure 1.5B. For these, if only genes in the top 50% of IMProve score were tested, 81% of the total positives would be captured. As noted before, this is a dramatic increase in efficiency. Excitingly, many of these families remain to be resolved structurally. Considering these results with the broader experimental data sets (Figure 1.3), no matter the number of proteins one is willing to test, the IMProve model enables selecting targets with a high probability of expression success in E. coli.
1.3.6 Sequence optimization for expression
The predictive performance of the model implies that the features defined here provide a coarse approximation of the fitness landscape for IMP expression. Attempting to optimize a single feature by modifying the sequence will likely affect the resulting score and expression due to changes in other features. Fluman, et al. provides an illustrative experiment (Fluman et al., 2014). For that work, it was hypothesized that altering the number of Shine-Dalgarno (SD)-like sites in the coding sequence of a IMP would affect expression level. To test this, silent mutations were engineered within the first 200 bases of three proteins (genes ygdD, brnQ, and ybjJ from E. coli) to increase the number of SD-like sites with the goal of improving expression. Expression trials demonstrated that only one of the proteins (BrnQ) had improved expression of folded protein. While the number of SD-like sites alone does not correlate, only 1 out of 3, the resulting changes in the IMProve score correlate with the changes in measured expression level, 3 out of 3 (Figure 1.5C). The IMProve model’s ability to capture the outcomes in this small test case illustrates the utility of integrating the contribution of the numerous parameters involved in IMP biogenesis.
1.4 Discussion
Here, we have demonstrated a statistically driven predictor, IMProve, that decodes from sequence the sum of biological features that drive expression, a feat not previously possible (Nørholm et al., 2013). The current best practice for characterization of an IMP target begins with the identification and testing of multiple homologs or variants for expression. The predictive power of IMProve enables this by providing a low barrier-to-entry method to enrich for positive outcomes by more than two-fold. IMProve allows for the prioritization of targets to test for expression making more optimal use of limited human and material resources. For groups with small scale projects such as individual labs, this means that for the same cost one would double the success rate. For large scale groups, such as companies or consortia, IMProve can reduce by half the cost required to obtain the same number of positive results. We provide the current predictor as a web service where scores can be calculated, and the method, associated data, and suggested analyses are publically available to catalyze progress across the community (clemonslab.caltech.edu).
Having shown that IMP expression can be predicted, the generalizability of the model is remarkable despite several known limitations that will be investigated in next generation predictors. Using data from a single study for training precluded including certain features that empirically influence expression level such as fusion tags and the context of the protein in an expression plasmid, e.g., the 5’ untranslated region, for which there was no variation in the Daley, Rapp, et al. dataset. Moreover, using a simple proof-of-concept linear model allowed for a straightforward and robust predictor; however, intrinsically it cannot be directly related to the biological underpinnings. While we can extract some biological inference, a linear combination of sequence features does not explicitly reflect the reality of physical limits for host cells. To some extent, constraint information is likely encoded in the complex architecture of the underlying sequence space (e.g., through the genetic code, TM prediction, RNA secondary structure analyses). Future statistical models that improve on these limitations will hone predictive power and more intricately characterize the interplay of variables that underlie IMP expression. By building upon the methodology here and by careful analysis of datasets in the literature, we expect to train IMP expression predictors for other host systems (e.g., eukaryotic cells).
A surprising outcome of our results is the observation of a quantitatively important contribution of the nucleotide sequence as a component of the IMProve score. This echoes the growing literature that aspects of the nucleotide sequence are important determinants of protein biogenesis in general (Chartron et al., 2016; Fluman et al., 2014; Gamble et al., 2016; Goodman et al., 2013; Li et al., 2012). While one expects that there may be different weights for various nucleotide derived features between soluble and IMPs, it is likely that these features are important for soluble proteins as well. An example of this is the importance of codon optimization for soluble protein expression, which has failed to show any general benefit for IMPs (Nørholm et al., 2012). Current expression predictors that have predictive power for soluble proteins have only used protein sequence for deriving the underlying feature set (Slabinski et al., 2007b; Wang et al., 2016). Future prediction methods will likely benefit from including nucleotide sequence features as done here.
The ability to predict phenotypic results using sequence based statistical models opens a variety of opportunities. As done here, this requires a careful understanding of the system and its underlying biological processes enumerated in a multitude of individual variables that impact the stated goal of the predictor, in this case enriching protein expression. As new features related to expression are discovered, future work will incorporate these leading to improved models. This can include features derived from other approaches such as the integration efficiency from coarse-grained molecular dynamics (Marshall et al., 2016; Niesen et al., 2017) and 5’ mRNA secondary structure/translation initiation efficiency (Goodman et al., 2013; Mirzadeh et al., 2015; Reis and Salis, 2020). Based on these results, expanding to new expression hosts such as eukaryotes seems entirely feasible, although a number of new features may need to be considered, e.g., glycosylation sites and trafficking signals. Moreover, the ability to score proteins for expressibility creates new avenues to computationally engineer IMPs for expression. The proof-of-concept described here required significant work to compile data from genomics consortia and the literature in a readily useable form. As data becomes more easily accessible, broadly leveraging diverse experimental outcomes to decode sequence-level information, an extension of this work, is anticipated.
1.5 Materials and Methods
Sequence mapping & retrieval and feature calculation was performed in Python 2.7 (Van Rossum and Drake Jr, 1995) using BioPython (Cock et al., 2009) and NumPy (van der Walt et al., 2011); executed and consolidated using Bash (shell) scripts; and parallelized where possible using GNU Parallel (Tange, 2011). Data analysis and presentation was done in R (R Core Team, 2015) within RStudio (RStudio Team, 2015) using magrittr (Bache and Wickham, 2014), plyr (Wickham, 2011), dplyr (Wickham and Francois, 2015), asbio (Aho, 2015), and datamart (Weinert, 2014) for data handling; ggplot2 (Wickham, 2016, p. 2), ggbeeswarm (Clarke and Sherrill-Mix, 2015), GGally (Schloerke et al., 2016), gridExtra (Auguie, 2015), cowplot (Wilke, 2015), scales (Wickham, 2015), viridis (Garnier, 2016), and RColorBrewer (Harrower and Brewer, 2003; Neuwirth, 2014) for plotting; multidplyr (Wickham, n.d.) with parallel (R Core Team, 2015) and foreach (Revolution Analytics and Weston, 2015a) with iterators (Revolution Analytics and Weston, 2015b) and doMC (Revolution Analytics and Weston, 2015c)/doParallel (Revolution Analytics and Weston, 2015d) for parallel processing; and roxygen2 (Wickham et al., 2015) for code organization and documentation as well as other packages as referenced.
1.5.1 Collection of data necessary for learning and evaluation
E. coli Sequence Data
The nucleotide sequences from (Daley et al., 2005) were deduced by reconstructing forward and reverse primers (i.e., ~20 nucleotide stretches) from each gene in Colibri (based on EcoGene 11), the original source cited and later verified these primers against an archival spreadsheet provided directly by Daniel Daley (personal communication). To account for sequence and annotation corrections made to the genome after Daley, Rapp, et al.’s work, these primers were directly used to reconstruct the amplified product from the most recent release of the E. coli K-12 substr. MG1655 genome (Zhou and Rudd, 2013) (EcoGene 3.0; U00096.3). Although Daniel Daley mentioned that raw reads from the Sanger sequencing runs may be available within his own archives, it was decided that the additional labor to retrieve this data and parse these reads would not significantly impact the model. The deduced nucleotide sequences were verified against the protein lengths given in Supplementary Table 1 from (Daley et al., 2005). The plasmid library tested in (Fluman et al., 2014) was provided by Daniel Daley, and those sequences are taken to be the same.
E. coli Training Data
The preliminary results using the mean-normalized activities echoed the findings of (Daley et al., 2005) that these do not correlate with sequence features either in the univariate sense (many simple linear regressions, Supplementary Table 1 (Daley et al., 2005)) or a multivariate sense (multiple linear regression, data not shown). This is presumably due to the loss of information regarding variability in expression level for given genes or due to the increase in variance of the normalized quantity (See Section 1.5.4) due to the normalization and averaging procedure. Daniel Daley and Mikaela Rapp provided spreadsheets of the outcomes from the 96-well plates used for their expression trials and sent scanned copies of the readouts from archival laboratory notebooks where the digital data was no longer accessible (personal communication). Those proteins without a reliable C-terminal localization (as given in the original work) or without raw expression outcomes were not included in further analyses.
Similarly, Nir Fluman also provided spreadsheets of the raw data from the set of three expression trials performed in (Fluman et al., 2014).
New York Consortium on Membrane Protein Structure (NYCOMPS) Data
Brian Kloss, Marco Punta, and Edda Kloppman provided a dataset of actions performed by the NYCOMPS center including expression outcomes in various conditions (Love et al., 2010; Punta et al., 2009). The protein sequences were mapped to NCBI GenInfo Identifier (GI) numbers either via the Entrez system (Schuler et al., 1996) or the Uniprot mapping service (UniProt Consortium, 2012). Each GI number was mapped to its nucleotide sequence via a combination of the NCBI Elink mapping service and the “coded_by” or “locus” tags of Coding Sequence (CDS) features within GenBank entries. Though custom-purpose code was written, a script from Peter Cock is available to do the same task via a similar mapping mechanism (Cock, 2009). To confirm all sequences, the TargetTrack (Chen et al., 2004) XML file was parsed for the internal NYCOMPS identifiers and compared for sequence identity to those that had been mapped using the custom script; 20 (less than 1%) of the sequences had minor inconsistencies and were manually replaced.
Archaeal transporters Data
The locus tags (“Gene Name” in Table 1) were mapped directly to the sequences and retrieved from NCBI (Ma et al., 2013). Pikyee Ma and Margarida Archer clarified questions regarding their work to inform the analysis.
GPCR Expression Data
Nucleotide sequences were collected by mapping the protein identifiers given in Table 1 from (Lundstrom et al., 2006) to protein GIs via the Uniprot mapping service (UniProt Consortium, 2012) and subsequently to their nucleotide sequences via the custom mapping script described above (see NYCOMPS 1.5.1.3). The sequence length and pI were validated against those provided. Renaud Wagner assisted in providing the nucleotide sequences for genes whose listed identifiers were unable to be mapped and/or did not pass the validation criteria as the MeProtDB (the sponsor of the GPCR project) does not provide a public archive.
Helicobacter pylori Data
Nucleotide sequences were retrieved by mapping the locus tags given in Supplemental Table 1 from (Psakis et al., 2007) to locus tags in the Jan 31, 2014 release of the H. pylori 26695 genome (AE000511.1). To verify sequence accuracy, sequences whose molecular weight matched that given by the authors were accepted. Those that did not match, in addition to the one locus tag that could not be mapped to the Jan 31, 2014 genome version, were retrieved from the Apr 9, 2015 release of the genome (NC_000915.1). Both releases are derived from the original sequencing project (Tomb et al., 1997). After this curation, all mapped sequences matched the reported molecular weight.
In this data set, expression tests were performed in three expression vectors and scored as 1, 2, or 3. Two vectors were scored via two methods. For these two vectors, the two scores were averaged to give a single number for the condition making them comparable to the third vector while yielding 2 additional thresholds (1.5 and 2.5) result in the 5 total curves shown (Figure 1.8B).
Mycobacterium tuberculosis Data
The authors note using TubercuList through GenoList (Lechat et al., 2008), therefore, nucleotide sequences were retrieved from the archival website based on the original sequencing project (Cole et al., 1998). The sequences corresponding to the identifiers and outcomes in Table 1 from (Korepanova et al., 2005) were validated against the provided molecular weight .
Secondary Transporter Data
GI Numbers given in Table 1 from (Surade et al., 2006) were matched to their CDS entries using the custom mapping script described above (see NYCOMPS 1.5.1.3). Only expression in E. coli with IPTG-inducible vectors was considered.
Thermotoga maratima Data
Gene names given in Table 1 (Dobrovetsky et al., 2005) were matched to CDS entries in the Jan 31, 2014 release of the Thermotoga maritima MSB8 genome (AE000512.1), a revised annotation of the original release (Nelson et al., 1999). The sequence length and molecular weight were validated against those provided.
Pseudomonas aeruginosa Data
Outcomes in Additional file 1 (Madhavan et al., 2010) were matched to coding sequences provided by Constance Jeffrey.
Shine-Dalgarno-like mutagenesis Data
Folded protein is quantified by densitometry measurement (Schindelin et al., 2012; Schneider et al., 2012) of the relevant band in Figure 6 of (Fluman et al., 2014). Relative difference is calculated as is standard (Equation 1.2):
\[ \frac{\text{metric}_\text{mutant} - \text{metric}_\text{wildtype}}{\frac{1}{2}\left| \text{metric}_\text{mutant} - \text{metric}_\text{wildtype} \right|} \tag{1.2}\]
1.5.2 Details related to the calculation of sequence features
Transmembrane segment topology was predicted using Phobius Constrained for the training data and Phobius for all other datasets (Käll et al., 2004). We were able to obtain the Phobius code and integrate it directly into our feature calculation pipeline resulting in significantly faster speeds than any other option. Several features were obtained by averaging per-site metrics (e.g., per-residue RONN3.2 disorder predictions) in windows of a specified length. Windowed tAI metrics are calculated over all 30 base windows (not solely over 10 codon windows). Table 1.1 includes an in-depth description of each feature. Future work will explore contributions of elements outside the gene of interest, e.g., ribosomal binding site, linkers, tags.
1.5.3 Preparation for model learning
Calculated sequence features for the IMPs in the E. coli dataset as well as raw activity measurements, i.e., each 96-well plate, were loaded into R. As is best practice in using Support Vector Machines, each feature was “centered” and “scaled” where the mean value of a given feature was subtracted from each data point and then divided by the standard deviation of that feature using preprocess (Kuhn, 2008). As is standard practice, the resulting set was then culled for those features of near zero-variance, over 95% correlation (Pearson’s r), and linear dependence (nearZeroVar, findCorrelation, findLinearCombos)(Kuhn, 2008). In particular this procedure removed extraneous degrees of freedom during the training process which carry little to no additional information with respect to the feature space and which may over represent certain redundant features. Features and outcomes for each list (“query”) were written into the SVMlight format using a modified svmlight.write (Weihs et al., 2005).
The final features were calculated for each sequence in the test datasets, prepared for scoring by “centering” and “scaling” by the training set parameters via preprocess (Kuhn, 2008), and then written into SVMlight format again using a modified svmlight.write.
1.5.4 Model selection, training, and evaluation using SVMrank
At the most basic level, our predictive model is a learned function that maps the parameter space (consisting of nucleotide and protein sequence features) to a response variable (expression level) through a set of governing weights (\(\vec{w}_1, \vec{w}_2, \ldots , \vec{w}_N\)). Depending on how the response variable is defined, these weights can be approximated using several different methods. As such, defining a response variable that is reflective of the available training data is key to selecting an appropriate learning algorithm.
The quantitative 96-well plate results (Daley et al., 2005) that comprise our training data do not offer an absolute expression metric valid over all plates—the top expressing proteins in one plate would not necessarily be the best expressing within another. As such, this problem is suited for preference-ranking methods. As a ranking problem, the response variable is the ordinal rank for each protein derived from its overexpression relative to the other members of the same plate of expression trials. In other words, the aim is to rank highly expressed proteins (based on numerous trials) at higher scores than lower expressed proteins by fitting against the order of expression outcomes from each constituent 96-well plate.
As the first work of this kind, the aim was to employ the simplest framework necessary taking in account the considerations above. The method chosen computes all valid pairwise classifications (i.e., within a single plate) transforming the original ranking problem into a binary classification problem. The algorithm outputs a score for each input by minimizing the number of swapped pairs thereby maximizing Kendall’s \(\tau\) (Kendall, 1938). For example, consider the following data generated via context \(A\): \((X_{A,1},\ Y_{A,1}), (X_{A,2},Y_{A,2})\) and \(B\): \((X_{B,1}, Y_{B,1}), (X_{B,2},Y_{B,2})\) where observed response follows as index \(i\), i.e., \(Y_{n} < Y_{n + 1}\). Binary classifier f (\(X_{i},\ X_{j})\) gives a score of 1 if an input pair matches its ordering criteria and \(- 1\) if not, i.e., \(Y_{i} < Y_{j}\):
\[ f\left( X_{A,1},X_{A,2} \right) = 1;f\left( X_{A,2},X_{A,1} \right) = - 1 \]
\[ f\left( X_{B,1},X_{B,2} \right) = 1;f\left( X_{B,2},X_{B,1} \right) = - 1 \]
\[ f\left( X_{A,1},X_{B,2} \right), f\left( X_{A,2},X_{B,1} \right) \text{ are invalid} \]
Free parameters describing \(f\) are calculated such that those calculated orderings \(f\left( X_{A,1} \right),f\left( X_{A,2} \right)\ldots;\ f\left( X_{B,1} \right),f\left( X_{B,2} \right)\ldots\) most closely agree (overall Kendall’s \(\tau\)) with the observed ordering \(\ Y_{n},\ Y_{n + 1},\ \ldots\). In this sense, \(f\) is a pairwise Learning to Rank method.
Within this class of models, a linear preference-ranking Support Vector Machine was employed (Joachims, 2002). To be clear, as an algorithm a preference-ranking SVM operates similarly to the canonical SVM binary classifier. In the traditional binary classification problem, a linear SVM seeks the maximally separating hyper-plane in the feature space between two classes, where class membership is determined by which side of the hyper-plane points reside. For some \(n\) linear separable training examples \(D = {\left\{ \ \left( x_{i} \right) \right|\ x_{i}\ \epsilon\ \mathbb{R}^{d}\}}^{n}\) and two classes \(y_{i}\ \epsilon\ \{ - 1,\ 1\}\), a linear SVM seeks a mapping from the d-dimensional feature space \(\mathbb{R}^{d} \rightarrow \{ - 1,\ 1\}\) by finding two maximally separated hyperplanes \(w\ \bullet x\ - b = 1\) and \(\ w\ \bullet x\ - b = - \ 1\) with constraints that \(w\ \bullet x_{i}\ - b \geq 1\ \)for all \(x_{i}\) with \(y_{i}\ \epsilon\ \{ 1\}\) and \(w\ \bullet x_{i}\ - b \leq \ - \ 1\) for all \(x_{i}\) with \(y_{i}\ \epsilon\ \{ - 1\}\). The feature weights correspond to the vector \(\vec{w}\), which is the vector perpendicular to the separating hyperplanes, and are computable in \(O(n\log{}n)\) implemented as part of the SVM^rank^ software package, though in \(O(n^2)\) (Tsochantaridis et al., 2005). See (Joachims, 2002) for an in-depth, technical discussion.
In a soft-margin SVM where training data is not linearly separable, a tradeoff between misclassified inputs and separation from the hyperplane must be specified. This parameter \(C\) was found by training models against raw data from Daley, Rapp, et al. with a grid of candidate \(C\) values (\(2^{n}\ \forall\ n\ \epsilon\ \lbrack - 5,\ \ 5\rbrack\)) and then evaluated against the raw “folded protein” measurements from Fluman, et al. The final model was chosen by selecting that with the lowest error from the process above (\(C = 2^5\)). To be clear, the final model is composed solely of a single weight for each feature; the tradeoff parameter \(C\) is only part of the training process.
Qualitatively, such a preference-ranking method constructs a model that ranks groups of proteins with higher expression level higher than other groups with lower expression value. In comparison to methods such as linear regression and binary classification, this approach is more robust and less affected by the inherent stochasticity of the training data.
1.5.5 Quantitative Assessment of Predictive Performance
In generating a predictive model, one aims to enrich for positive outcomes while ensuring they do not come at the cost of increased false positive diagnoses. This is formalized in Receiver Operating Characteristic (ROC) theory (for a primer see (Swets et al., 2000)), where the true positive rate is plotted against the false positive rate for all classification thresholds (score cutoffs in the ranked list). In this framework, the overall ability of the model to resolve positive from negative outcomes is evaluated by analyzing the Area Under a ROC curve (AUC) where AUCperfect=100% and AUCrandom=50% (percentage signs are omitted throughout the text and figures). All ROCs are calculated through pROC (Robin et al., 2011) using the analytic Delong method for AUC confidence intervals (DeLong et al., 1988). Bootstrapped AUC CIs (\(N = 10^6\)) were precise to 4 decimal places suggesting that analytic CIs are valid for the NYCOMPS dataset.
With several of our datasets, no definitive standard or clear-cut classification for positive expression exists. However, the aim is to show and test all reasonable classification thresholds of positive expression for each dataset in order to evaluate predictive performance as follows:
Training data
The outcomes are quantitative (activity level), so each ROC is calculated by normalizing within each dataset to the standard well subject to the discussion in 4a above (LepB for PhoA, and InvLepB for GFP) (examples in Figure 1.1D) for each possible threshold, i.e., each normalized expression value with each AUC plotted in Figure 1.1E. 95% confidence intervals of Spearman’s \(\rho\) are given by 106 iterations of a bias-corrected and accelerated (BCa) bootstrap of the data (Figure 1.1A,C) (Canty and Ripley, 2015).
Large-scale
ROCs were calculated for each of the expression classes (Figure 1.2E). Regardless of the split, predictive performance is noted. The binwidth for the histogram was determined using the Freedman-Diaconis rule(Freedman and Diaconis, 1981), and scores outside the plotted range comprising <0.6% of the density were implicitly hidden.
Small-scale
Classes can be defined in many different ways. To be principled about the matter, ROCs for each possible cutoff are presented based on definitions from each publication (Figure 1.3C,E,G; Figure 1.8B,D,F). See Section 1.5.1 for any necessary details about outcome classifications for each dataset.
1.5.6 Feature Weights
Weights for the learned SVM are pulled directly from the model file produced by SVMlight and are given in Table 1.1.
1.5.7 Availability
All analysis is documented in a series of R notebooks (Xie, 2014) available openly at github.com/clemlab/ml-ecoli-IMProve. These notebooks provide fully executable instructions for the reproduction of the analyses and the generation of figures and statistics in this study. The IMProve model is available as a web service at clemonslab.caltech.edu.
1.6 Acknowledgements
We thank Daniel Daley and Thomas Miller’s group for discussion, Yaser Abu-Mostafa and Yisong Yue for guidance regarding machine learning, Niles Pierce for providing NUPACK source code (Zadeh et al., 2011), Welison Floriano and Naveed Near-Ansari for maintaining local computing resources, and Samuel Schulte for suggesting the model’s name. We thank Michiel Niesen, Stephen Marshall, Thomas Miller, Reid van Lehn, James Bowie, and Tom Rapoport for comments on the manuscript. Models and analyses are possible thanks to raw experimental data provided by Daniel Daley and Mikaela Rapp (Daley et al., 2005); Nir Fluman (Fluman et al., 2014); Edda Kloppmann, Brian Kloss, and Marco Punta from NYCOMPS (Love et al., 2010; Punta et al., 2009); Pikyee Ma (Ma et al., 2013); Renaud Wagner (Lundstrom et al., 2006); Florent Bernaudat (Bernaudat et al., 2011), and Constance Jeffrey (Madhavan et al., 2010).
We acknowledge funding from an NIH Pioneer Award to WMC (5DP1GM105385); a Benjamin M. Rosen graduate fellowship, a NIH/NRSA training grant (5T32GM07616), and a NSF Graduate Research fellowship to SMS; and an Arthur A. Noyes Summer Undergraduate Research Fellowship to NJ. Computational time was provided by Stephen Mayo and Douglas Rees. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. 1144469. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1053575 (Towns et al., 2014).
1.7 Supplementary Figures

1.8 Supplementary Tables

References
Abell BM, Mullen RT. 2011. Tail-anchored membrane proteins: Exploring
the complex diversity of tail-anchored-protein targeting in plant cells.
Plant Cell Rep 30:137–151. doi:10.1007/s00299-010-0925-6
Aho K. 2015. Asbio:
A collection of statistical tools for biologists.
Almagro Armenteros JJ, Salvatore M, Emanuelsson O, Winther O, von Heijne
G, Elofsson A, Nielsen H. 2019. Detecting sequence signals in targeting
peptides using deep learning. Life Sci Alliance
2:e201900429. doi:10.26508/lsa.201900429
Almagro Armenteros JJ, Sønderby CK, Sønderby SK, Nielsen H, Winther O.
2017. DeepLoc: Prediction of protein subcellular
localization using deep learning. Bioinformatics
33:3387–3395. doi:10.1093/bioinformatics/btx431
Anderson SA, Satyanarayan MB, Wessendorf RL, Lu Y, Fernandez DE. 2021. A
homolog of GuidedEntry of Tail-anchored Proteins3 functions in
membrane-specific protein targeting in chloroplasts of
Arabidopsis. Plant Cell
33:2812–2833. doi:10.1093/plcell/koab145
Anderson SA, Singhal R, Fernandez DE. 2019. Membrane-Specific
Targeting of Tail-Anchored Proteins SECE1 and
SECE2 Within Chloroplasts. Front Plant Sci
10:1401. doi:10.3389/fpls.2019.01401
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis
AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L,
Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM,
Sherlock G. 2000. Gene ontology: Tool for the unification of biology.
The Gene Ontology Consortium. Nat Genet
25:25–29. doi:10.1038/75556
Asseck LY, Mehlhorn DG, Monroy JR, Ricardi MM, Breuninger H, Wallmeroth
N, Berendzen KW, Nowrousian M, Xing S, Schwappach B, Bayer M, Grefen C.
2021. Endoplasmic reticulum membrane receptors of the GET
pathway are conserved throughout eukaryotes. Proc Natl Acad Sci U S
A 118:e2017636118. doi:10.1073/pnas.2017636118
Auguie B. 2015. gridExtra: Miscellaneous functions
for "Grid" graphics.
Aurora R, Rose GD. 1998. Helix capping. Protein Sci
7:21–38. doi:10.1002/pro.5560070103
Aviram N, Ast T, Costa EA, Arakel EC, Chuartzman SG, Jan CH,
Haßdenteufel S, Dudek J, Jung M, Schorr S, Zimmermann R, Schwappach B,
Weissman JS, Schuldiner M. 2016. The SND proteins
constitute an alternative targeting route to the endoplasmic reticulum.
Nature 540:134–138. doi:10.1038/nature20169
Aviram N, Schuldiner M. 2017. Targeting and translocation of proteins to
the endoplasmic reticulum at a glance. J Cell Sci
130:4079–4085. doi:10.1242/jcs.204396
Bache SM, Wickham H. 2014. Magrittr:
A forward-pipe operator for R.
Bédard J, Kubis S, Bimanadham S, Jarvis P. 2007. Functional similarity
between the chloroplast translocon component, Tic40, and
the human co-chaperone, Hsp70-interacting
protein (Hip). J Biol Chem
282:21404–21414. doi:10.1074/jbc.M611545200
Bédard J, Trösch R, Wu F, Ling Q, Flores-Pérez Ú, Töpel M, Nawaz F,
Jarvis P. 2017. Suppressors of the Chloroplast Protein Import
Mutant Tic40 Reveal a Genetic Link
between Protein Import and Thylakoid
Biogenesis. Plant Cell 29:1726–1747.
doi:10.1105/tpc.16.00962
Beilharz T, Egan B, Silver PA, Hofmann K, Lithgow T. 2003. Bipartite
signals mediate subcellular targeting of tail-anchored membrane proteins
in Saccharomyces cerevisiae. J Biol Chem
278:8219–8223. doi:10.1074/jbc.M212725200
Berardini TZ, Reiser L, Li D, Mezheritsky Y, Muller R, Strait E, Huala
E. 2015. The Arabidopsis information resource:
Making and mining the "gold standard" annotated reference
plant genome. Genesis 53:474–485. doi:10.1002/dvg.22877
Berger SA, Krompass D, Stamatakis A. 2011. Performance, accuracy, and
Web server for evolutionary placement of short sequence
reads under maximum likelihood. Syst Biol
60:291–302. doi:10.1093/sysbio/syr010
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H,
Shindyalov IN, Bourne PE. 2000. The protein data bank. Nucleic Acids
Research 28:235–242.
Bernaudat F, Frelet-Barrand A, Pochon N, Dementin S, Hivin P, Boutigny
S, Rioux J-B, Salvi D, Seigneurin-Berny D, Richaud P, Joyard J, Pignol
D, Sabaty M, Desnos T, Pebay-Peyroula E, Darrouzet E, Vernet T, Rolland
N. 2011. Heterologous expression of membrane proteins: Choosing the
appropriate host. PloS One 6:e29191. doi:10.1371/journal.pone.0029191
Bernstein HD, Poritz MA, Strub K, Hoben PJ, Brenner S, Walter P. 1989.
Model for signal sequence recognition from amino-acid sequence of
54K subunit of signal recognition particle. Nature
340:482–486. doi:10.1038/340482a0
Bertone P, Kluger Y, Lan N, Zheng D, Christendat D, Yee A, Edwards AM,
Arrowsmith CH, Montelione GT, Gerstein M. 2001. SPINE: An
integrated tracking database and data mining approach for identifying
feasible targets in high-throughput structural proteomics. Nucleic
Acids Research 29:2884–2898.
Biebl MM, Buchner J. 2019. Structure, Function, and
Regulation of the Hsp90 Machinery. Cold
Spring Harb Perspect Biol 11:a034017. doi:10.1101/cshperspect.a034017
Bill RM, Henderson PJF, Iwata S, Kunji ERS, Michel H, Neutze R, Newstead
S, Poolman B, Tate CG, Vogel H. 2011. Overcoming barriers to membrane
protein structure determination. Nature Biotechnology
29:335–340. doi:10.1038/nbt.1833
Blum M, Chang H-Y, Chuguransky S, Grego T, Kandasaamy S, Mitchell A,
Nuka G, Paysan-Lafosse T, Qureshi M, Raj S, Richardson L, Salazar GA,
Williams L, Bork P, Bridge A, Gough J, Haft DH, Letunic I,
Marchler-Bauer A, Mi H, Natale DA, Necci M, Orengo CA, Pandurangan AP,
Rivoire C, Sigrist CJA, Sillitoe I, Thanki N, Thomas PD, Tosatto SCE, Wu
CH, Bateman A, Finn RD. 2021. The InterPro protein families
and domains database: 20 years on. Nucleic Acids Res
49:D344–D354. doi:10.1093/nar/gkaa977
Bodensohn US, Simm S, Fischer K, Jäschke M, Groß LE, Kramer K, Ehmann C,
Rensing SA, Ladig R, Schleiff E. 2019. The intracellular distribution of
the components of the GET system in vascular plants.
Biochim Biophys Acta Mol Cell Res
1866:1650–1662. doi:10.1016/j.bbamcr.2019.06.012
Bois J. 2020. Justinbois/Bebi103: 0.1.0. doi:10.22002/D1.1615
Borgese N, Colombo S, Pedrazzini E. 2003. The tale of tail-anchored
proteins: Coming from the cytosol and looking for a membrane. J Cell
Biol 161:1013–1019. doi:10.1083/jcb.200303069
Borkin MA, Gajos KZ, Peters A, Mitsouras D, Melchionna S, Rybicki FJ,
Feldman CL, Pfister H. 2011. Evaluation of artery visualizations for
heart disease diagnosis. IEEE Trans Vis Comput Graph
17:2479–2488. doi:10.1109/TVCG.2011.192
Borland D, Taylor RM II. 2007. Rainbow Color Map
(Still) Considered Harmful. IEEE Comput
Grap Appl 27:14–17. doi:10.1109/MCG.2007.323435
Bradley P, Misura KMS, Baker D. 2005. Toward high-resolution de novo
structure prediction for small proteins. Science
309:1868–1871. doi:10.1126/science.1113801
Buchanan G, Ricciardelli C, Harris JM, Prescott J, Yu ZC-L, Jia L,
Butler LM, Marshall VR, Scher HI, Gerald WL, Coetzee GA, Tilley WD.
2007. Control of androgen receptor signaling in prostate cancer by the
cochaperone small glutamine rich tetratricopeptide repeat containing
protein alpha. Cancer Res 67:10087–10096.
doi:10.1158/0008-5472.CAN-07-1646
Burri L, Lithgow T. 2004. A complete set of SNAREs in
yeast. Traffic 5:45–52. doi:10.1046/j.1600-0854.2003.00151.x
Callahan MA, Handley MA, Lee YH, Talbot KJ, Harper JW, Panganiban AT.
1998. Functional interaction of human immunodeficiency virus type 1
Vpu and Gag with a novel member of the
tetratricopeptide repeat protein family. J Virol
72:5189–5197. doi:10.1128/JVI.72.6.5189-5197.1998
Canty A, Ripley BD. 2015. Boot: Bootstrap R (S-plus) functions.
Caplan AJ. 2003. What is a co-chaperone? Cell Stress Chaperones
8:105–107. doi:10.1379/1466-1268(2003)008<0105:wiac>2.0.co;2
Chang HC, Nathan DF, Lindquist S. 1997. In vivo analysis of the
Hsp90 cochaperone Sti1 (P60). Mol Cell
Biol 17:318–325. doi:10.1128/MCB.17.1.318
Chartron JW, Clemons WM, Suloway CJM. 2012a. The complex process of
GETting tail-anchored membrane proteins to the
ER. Curr Opin Struct Biol
22:217–224. doi:10.1016/j.sbi.2012.03.001
Chartron JW, Gonzalez GM, Clemons WM. 2011. A structural model of the
Sgt2 protein and its interactions with chaperones and the
Get4/Get5 complex. J Biol Chem
286:34325–34334. doi:10.1074/jbc.M111.277798
Chartron JW, Hunt KCL, Frydman J. 2016. Cotranslational
signal-independent SRP preloading during membrane
targeting. Nature 536:224–228. doi:10.1038/nature19309
Chartron JW, VanderVelde DG, Clemons WM. 2012b. Structures of the
Sgt2/SGTA dimerization domain with the
Get5/UBL4A UBL domain reveal an interaction
that forms a conserved dynamic interface. Cell Rep
2:1620–1632. doi:10.1016/j.celrep.2012.10.010
Chen CM, Misra TK, Silver S, Rosen BP. 1986. Nucleotide sequence of the
structural genes for an anion pump. The plasmid-encoded
arsenical resistance operon. J Biol Chem
261:15030–15038.
Chen L, Oughtred R, Berman HM, Westbrook J. 2004. TargetDB:
A target registration database for structural genomics projects.
Bioinformatics 20:2860–2862. doi:10.1093/bioinformatics/bth300
Chen S, Smith DF. 1998. Hop as an adaptor in the heat shock protein 70
(Hsp70) and Hsp90 chaperone machinery. J Biol Chem
273:35194–35200. doi:10.1074/jbc.273.52.35194
Chio US, Cho H, Shan S-O. 2017. Mechanisms of Tail-Anchored
Membrane Protein Targeting and Insertion. Annu
Rev Cell Dev Biol 33:417–438. doi:10.1146/annurev-cellbio-100616-060839
Chio US, Chung S, Weiss S, Shan S-O. 2019. A Chaperone Lid Ensures
Efficient and Privileged Client Transfer during
Tail-Anchored Protein Targeting. Cell Rep
26:37–44.e7. doi:10.1016/j.celrep.2018.12.035
Chitwood PJ, Juszkiewicz S, Guna A, Shao S, Hegde RS. 2018. EMC Is
Required to Initiate Accurate Membrane Protein
Topogenesis. Cell 175:1507–1519.e16.
doi:10.1016/j.cell.2018.10.009
Cho H, Shan S-O. 2018. Substrate relay in an Hsp70-cochaperone cascade safeguards tail-anchored
membrane protein targeting. EMBO J 37:e99264.
doi:10.15252/embj.201899264
Choi H, Yi T, Ha S-H. 2021. Diversity of Plastid Types and
Their Interconversions. Front Plant Sci
12:692024. doi:10.3389/fpls.2021.692024
Chou M-L, Chu C-C, Chen L-J, Akita M, Li H. 2006. Stimulation of
transit-peptide release and ATP hydrolysis by a cochaperone
during protein import into chloroplasts. J Cell Biol
175:893–900. doi:10.1083/jcb.200609172
Chou M-L, Fitzpatrick LM, Tu S-L, Budziszewski G, Potter-Lewis S, Akita
M, Levin JZ, Keegstra K, Li H-M. 2003. Tic40, a membrane-anchored
co-chaperone homolog in the chloroplast protein translocon. EMBO
J 22:2970–2980. doi:10.1093/emboj/cdg281
Chu R, Takei J, Knowlton JR, Andrykovitch M, Pei W, Kajava AV, Steinbach
PJ, Ji X, Bai Y. 2002. Redesign of a four-helix bundle protein by phage
display coupled with proteolysis and structural characterization by
NMR and X-ray crystallography.
J Mol Biol 323:253–262. doi:10.1016/s0022-2836(02)00884-7
Chun E, Thompson AA, Liu W, Roth CB, Griffith MT, Katritch V, Kunken J,
Xu F, Cherezov V, Hanson MA, Stevens RC. 2012. Fusion partner toolchest
for the stabilization and crystallization of G
protein-coupled receptors. Structure
20:967–976. doi:10.1016/j.str.2012.04.010
Clarke E, Sherrill-Mix S. 2015. Ggbeeswarm:
Categorical scatter (violin point) plots.
Clemons WM, Gowda K, Black SD, Zwieb C, Ramakrishnan V. 1999. Crystal
structure of the conserved subdomain of human protein
SRP54M at 2.1 A resolution: Evidence for the
mechanism of signal peptide binding. J Mol Biol
292:697–705. doi:10.1006/jmbi.1999.3090
Cock P. 2009. [BioPython]
Downloading CDS sequences.
Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I,
Hamelryck T, Kauff F, Wilczynski B, de Hoon MJL. 2009. Biopython: Freely
available Python tools for computational molecular biology
and bioinformatics. Bioinformatics
25:1422–1423. doi:10.1093/bioinformatics/btp163
Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon
SV, Eiglmeier K, Gas S, Barry CE, Tekaia F, Badcock K, Basham D, Brown
D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S,
Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S,
Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter
S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K,
Whitehead S, Barrell BG. 1998. Deciphering the biology of
Mycobacterium tuberculosis from the complete genome
sequence. Nature 393:537–544. doi:10.1038/31159
Coleman JR, Papamichail D, Skiena S, Futcher B, Wimmer E, Mueller S.
2008. Virus attenuation by genome-scale changes in codon pair bias.
Science (New York, NY) 320:1784–1787. doi:10.1126/science.1155761
Conklin D, Holderman S, Whitmore TE, Maurer M, Feldhaus AL. 2000.
Molecular cloning, chromosome mapping and characterization of
UBQLN3 a testis-specific gene that contains an
ubiquitin-like domain. Gene 249:91–98. doi:10.1016/s0378-1119(00)00122-0
Costello JL, Castro IG, Camões F, Schrader TA, McNeall D, Yang J,
Giannopoulou E-A, Gomes S, Pogenberg V, Bonekamp NA, Ribeiro D, Wilmanns
M, Jedd G, Islinger M, Schrader M. 2017. Predicting the targeting of
tail-anchored proteins to subcellular compartments in mammalian cells.
J Cell Sci 130:1675–1687. doi:10.1242/jcs.200204
Coto ALS, Seraphim TV, Batista FAH, Dores-Silva PR, Barranco ABF,
Teixeira FR, Gava LM, Borges JC. 2018. Structural and functional studies
of the Leishmania braziliensis SGT
co-chaperone indicate that it shares structural features with
HIP and can interact with both Hsp90 and
Hsp70 with similar affinities. Int J Biol Macromol
118:693–706. doi:10.1016/j.ijbiomac.2018.06.123
Cziepluch C, Kordes E, Poirey R, Grewenig A, Rommelaere J, Jauniaux JC.
1998. Identification of a novel cellular TPR-containing protein, SGT, that
interacts with the nonstructural protein NS1 of parvovirus
H-1. J Virol 72:4149–4156. doi:10.1128/JVI.72.5.4149-4156.1998
da Fonseca ACC, Matias D, Geraldo LHM, Leser FS, Pagnoncelli I, Garcia
C, do Amaral RF, da Rosa BG, Grimaldi I, de Camargo Magalhães ES,
Cóppola-Segovia V, de Azevedo EM, Zanata SM, Lima FRS. 2021. The
multiple functions of the co-chaperone stress inducible protein 1.
Cytokine Growth Factor Rev 57:73–84. doi:10.1016/j.cytogfr.2020.06.003
Daley DO, Rapp M, Granseth E, Melén K, Drew D, von Heijne G. 2005.
Global topology analysis of the Escherichia coli inner
membrane proteome. Science (New York, NY)
308:1321–1323. doi:10.1126/science.1109730
Dantuma NP, Heinen C, Hoogstraten D. 2009. The ubiquitin receptor
Rad23: At the crossroads of nucleotide excision repair and
proteasomal degradation. DNA Repair (Amst)
8:449–460. doi:10.1016/j.dnarep.2009.01.005
Darby JF, Krysztofinska EM, Simpson PJ, Simon AC, Leznicki P,
Sriskandarajah N, Bishop DS, Hale LR, Alfano C, Conte MR,
Martínez-Lumbreras S, Thapaliya A, High S, Isaacson RL. 2014. Solution
structure of the SGTA dimerisation domain and investigation
of its interactions with the ubiquitin-like domains of BAG6
and UBL4A. PLoS One 9:e113281.
doi:10.1371/journal.pone.0113281
DeLong ER, DeLong DM, Clarke-Pearson DL. 1988. Comparing the areas under
two or more correlated receiver operating characteristic curves: A
nonparametric approach. Biometrics 44:837–845.
Deng H-X, Chen W, Hong S-T, Boycott KM, Gorrie GH, Siddique N, Yang Y,
Fecto F, Shi Y, Zhai H, Jiang H, Hirano M, Rampersaud E, Jansen GH,
Donkervoort S, Bigio EH, Brooks BR, Ajroud K, Sufit RL, Haines JL,
Mugnaini E, Pericak-Vance MA, Siddique T. 2011. Mutations in
UBQLN2 cause dominant X-linked
juvenile and adult-onset ALS and ALS/dementia.
Nature 477:211–215. doi:10.1038/nature10353
Denic V. 2012. A portrait of the GET pathway as a
surprisingly complicated young man. Trends Biochem Sci
37:411–417. doi:10.1016/j.tibs.2012.07.004
Dobrogojski J, Adamiec M, Luciński R. 2020. The chloroplast genome: A
review. Acta Physiol Plant 42:98. doi:10.1007/s11738-020-03089-x
Dobrovetsky E, Lu ML, Andorn-Broza R, Khutoreskaya G, Bray JE, Savchenko
A, Arrowsmith CH, Edwards AM, Koth CM. 2005. High-throughput production
of prokaryotic membrane proteins. Journal of Structural and
Functional Genomics 6:33–50. doi:10.1007/s10969-005-1363-5
dos Reis M, Wernisch L, Savva R. 2003. Unexpected correlations between
gene expression and codon usage bias from microarray data for the whole
Escherichia coli K-12 genome. Nucleic
Acids Research 31:6976–6985.
Droettboom M. 2016. Change
default color map (Fix matplotlib#875).
Drozdetskiy A, Cole C, Procter J, Barton GJ. 2015. JPred4:
A protein secondary structure prediction server. Nucleic Acids
Res 43:W389–394. doi:10.1093/nar/gkv332
Duncan O, van der Merwe MJ, Daley DO, Whelan J. 2013. The outer
mitochondrial membrane in higher plants. Trends Plant Sci
18:207–217. doi:10.1016/j.tplants.2012.12.004
Dupzyk A, Williams JM, Bagchi P, Inoue T, Tsai B. 2017.
SGTA-Dependent Regulation of Hsc70 Promotes Cytosol
Entry of Simian Virus 40 from the Endoplasmic
Reticulum. J Virol 91:e00232–17. doi:10.1128/JVI.00232-17
Dutta S, Tan Y-J. 2008. Structural and functional characterization of
human SGT and its interaction with Vpu of the
human immunodeficiency virus type 1. Biochemistry
47:10123–10131. doi:10.1021/bi800758a
Dyson HJ, Wright PE. 2004. Unfolded proteins and protein folding studied
by NMR. Chem Rev 104:3607–3622.
doi:10.1021/cr030403s
Eddins S. 2014. A New Colormap for MATLAB –
Part 1 – Introduction. https://blogs.mathworks.com/steve/2014/10/13/a-new-colormap-for-matlab-part-1-introduction/
Eddy SR, Wheeler TJ. 2020. The HMMER development team.
HMMER user’s guide: biological sequence analysis using profile
hidden markov models.
Eisenberg D, Schwarz E, Komaromy M, Wall R. 1984. Analysis of membrane
and surface protein sequences with the hydrophobic moment plot. J
Mol Biol 179:125–142. doi:10.1016/0022-2836(84)90309-7
El Ayadi A, Stieren ES, Barral JM, Boehning D. 2013. Ubiquilin-1 and
protein quality control in Alzheimer disease.
Prion 7:164–169. doi:10.4161/pri.23711
El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi
M, Richardson LJ, Salazar GA, Smart A, Sonnhammer ELL, Hirsh L, Paladin
L, Piovesan D, Tosatto SCE, Finn RD. 2019. The Pfam protein
families database in 2019. Nucleic Acids Res
47:D427–D432. doi:10.1093/nar/gky995
Elsasser S, Gali RR, Schwickart M, Larsen CN, Leggett DS, Müller B, Feng
MT, Tübing F, Dittmar GAG, Finley D. 2002. Proteasome subunit
Rpn1 binds ubiquitin-like protein domains. Nat Cell
Biol 4:725–730. doi:10.1038/ncb845
Engelman DM, Steitz TA, Goldman A. 1986. Identifying nonpolar
transbilayer helices in amino acid sequences of membrane proteins.
Annual Review of Biophysics and Biophysical Chemistry
15:321–353. doi:10.1146/annurev.bb.15.060186.001541
Eshaghi S, Hedrén M, Nasser MIA, Hammarberg T, Thornell A, Nordlund P.
2005. An efficient strategy for high-throughput expression screening of
recombinant integral membrane proteins. Protein Science
14:676–683. doi:10.1110/ps.041127005
Fan G-H, Yang W, Sai J, Richmond A. 2002. Hsc/Hsp70
interacting protein (hip) associates with CXCR2 and
regulates the receptor signaling and trafficking. J Biol Chem
277:6590–6597. doi:10.1074/jbc.M110588200
Fauchere J, Pliska V. 1983. Hydrophobic parameters II of
amino acid side-chains from the partitioning of N-acetyl-amino acid amides. Eur J Med
Chem 18.
Fernandez-Patron C, Hardy E, Sosa A, Seoane J, Castellanos L. 1995.
Double staining of coomassie blue-stained polyacrylamide gels by
imidazole-sodium dodecyl sulfate-zinc reverse staining: Sensitive
detection of coomassie blue-undetected proteins. Anal Biochem
224:263–269. doi:10.1006/abio.1995.1039
Figueiredo Costa B, Cassella P, Colombo SF, Borgese N. 2018.
Discrimination between the endoplasmic reticulum and mitochondria by
spontaneously inserting tail-anchored proteins. Traffic
19:182–197. doi:10.1111/tra.12550
Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger
A, Hetherington K, Holm L, Mistry J, Sonnhammer ELL, Tate J, Punta M.
2014. Pfam: The protein families database. Nucleic Acids
Research 42:D222–230. doi:10.1093/nar/gkt1223
Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter
SC, Punta M, Qureshi M, Sangrador-Vegas A, Salazar GA, Tate J, Bateman
A. 2016. The Pfam protein families database: Towards a more
sustainable future. Nucleic Acids Res
44:D279–285. doi:10.1093/nar/gkv1344
Fishbain S, Prakash S, Herrig A, Elsasser S, Matouschek A. 2011. Rad23
escapes degradation because it lacks a proteasome initiation region.
Nat Commun 2:192. doi:10.1038/ncomms1194
Fluman N, Navon S, Bibi E, Pilpel Y. 2014. mRNA-programmed translation pauses in the
targeting of E. Coli membrane proteins. eLife
3. doi:10.7554/eLife.03440
Fowler DM, Fields S. 2014. Deep mutational scanning: A new style of
protein science. Nat Methods 11:801–807.
doi:10.1038/nmeth.3027
Frain KM, Gangl D, Jones A, Zedler JAZ, Robinson C. 2016. Protein
translocation and thylakoid biogenesis in cyanobacteria. Biochim
Biophys Acta 1857:266–273. doi:10.1016/j.bbabio.2015.08.010
Franklin M. 2013. [Phenixbb]
coot and mtz interpretation.
Freedman D, Diaconis P. 1981. On the histogram as a density
estimator:L 2 theory. Zeitschrift für
Wahrscheinlichkeitstheorie und Verwandte Gebiete
57:453–476. doi:10.1007/BF01025868
Fry MY, Clemons WM. 2018. Complexity in targeting membrane proteins.
Science 359:390–391. doi:10.1126/science.aar5992
Fry MY, Najdrová V, Maggiolo AO, Saladi SM, Doležal P, Clemons WM. 2022.
Structurally derived universal mechanism for the catalytic cycle of the
tail-anchored targeting factor Get3. Nat Struct Mol
Biol 29:820–830. doi:10.1038/s41594-022-00798-4
Fry MY, Saladi SM, Cunha A, Clemons WM. 2021. Sequence-based features
that are determinant for tail-anchored membrane protein sorting in
eukaryotes. Traffic 22:306–318. doi:10.1111/tra.12809
Gabanyi MJ, Adams PD, Arnold K, Bordoli L, Carter LG, Flippen-Andersen
J, Gifford L, Haas J, Kouranov A, McLaughlin WA, Micallef DI, Minor W,
Shah R, Schwede T, Tao Y-P, Westbrook JD, Zimmerman M, Berman HM. 2011.
The Structural Biology Knowledgebase: A portal to protein
structures, sequences, functions, and methods. Journal of Structural
and Functional Genomics 12:45–54. doi:10.1007/s10969-011-9106-2
Gamble CE, Brule CE, Dean KM, Fields S, Grayhack EJ. 2016. Adjacent
codons act in concert to modulate translation efficiency in yeast.
Cell 166:679–690. doi:10.1016/j.cell.2016.05.070
Garnier S. 2016. Viridis:
Default color maps from ’matplotlib’.
Gautier R, Douguet D, Antonny B, Drin G. 2008. HELIQUEST: A
web server to screen sequences with specific alpha-helical properties.
Bioinformatics 24:2101–2102. doi:10.1093/bioinformatics/btn392
Geertsma ER, Groeneveld M, Slotboom D-J, Poolman B. 2008. Quality
control of overexpressed membrane proteins. Proceedings of the
National Academy of Sciences of the United States of America
105:5722–5727. doi:10.1073/pnas.0802190105
Gelvin SB. 2017. Integration of Agrobacterium T-DNA into
the Plant Genome. Annu Rev Genet
51:195–217. doi:10.1146/annurev-genet-120215-035320
Gene Ontology Consortium. 2021. The Gene Ontology resource:
Enriching a GOld mine. Nucleic Acids Res
49:D325–D334. doi:10.1093/nar/gkaa1113
Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, Smith HO. 2009.
Enzymatic assembly of DNA molecules up to several hundred
kilobases. Nat Methods 6:343–345. doi:10.1038/nmeth.1318
Goodman DB, Church GM, Kosuri S. 2013. Causes and effects of N-terminal codon bias in bacterial genes.
Science (New York, NY) 342:475–479. doi:10.1126/science.1241934
Gordon E, Horsefield R, Swarts HGP, de Pont JJHHM, Neutze R, Snijder A.
2008. Effective high-throughput overproduction of membrane proteins in
Escherichia coli. Protein Expression and
Purification 62:1–8. doi:10.1016/j.pep.2008.07.005
Graether SP. 2018. Troubleshooting Guide to
Expressing Intrinsically Disordered Proteins for
Use in NMR Experiments. Front Mol
Biosci 5:118. doi:10.3389/fmolb.2018.00118
Gristick HB, Rao M, Chartron JW, Rome ME, Shan S-O, Clemons WM. 2014.
Crystal structure of ATP-bound
Get3-Get4-Get5 complex reveals regulation of Get3 by
Get4. Nat Struct Mol Biol
21:437–442. doi:10.1038/nsmb.2813
Guna A, Hegde RS. 2018. Transmembrane Domain Recognition
during Membrane Protein Biogenesis and Quality
Control. Curr Biol 28:R498–R511. doi:10.1016/j.cub.2018.02.004
Guna A, Volkmar N, Christianson JC, Hegde RS. 2018. The ER
membrane protein complex is a transmembrane domain insertase.
Science 359:470–473. doi:10.1126/science.aao3099
Hara T, Kamura T, Kotoshiba S, Takahashi H, Fujiwara K, Onoyama I,
Shirakawa M, Mizushima N, Nakayama KI. 2005. Role of the
UBL-UBA protein KPC2 in degradation of P27 at
G1 phase of the cell cycle. Mol Cell Biol
25:9292–9303. doi:10.1128/MCB.25.21.9292-9303.2005
Harrower M, Brewer CA. 2003. ColorBrewer.org: An online
tool for selecting colour schemes for maps. Cartographic Journal,
The 40:27–37.
Hartley AM, Lukoyanova N, Zhang Y, Cabrera-Orefice A, Arnold S, Meunier
B, Pinotsis N, Maréchal A. 2019. Structure of yeast cytochrome c oxidase
in a supercomplex with cytochrome Bc1. Nat Struct Mol Biol
26:78–83. doi:10.1038/s41594-018-0172-z
Hegde RS, Keenan RJ. 2022. The mechanisms of integral membrane protein
biogenesis. Nat Rev Mol Cell Biol 23:107–124.
doi:10.1038/s41580-021-00413-2
Hegde RS, Keenan RJ. 2011. Tail-anchored membrane protein insertion into
the endoplasmic reticulum. Nat Rev Mol Cell Biol
12:787–798. doi:10.1038/nrm3226
Hendrickson WA. 2016. Atomic-level analysis of membrane-protein
structure. Nature Structural & Molecular Biology
23:464–467. doi:10.1038/nsmb.3215
Hessa T, Meindl-Beinker NM, Bernsel A, Kim H, Sato Y, Lerch-Bader M,
Nilsson I, White SH, von Heijne G. 2007. Molecular code for
transmembrane-helix recognition by the Sec61 translocon.
Nature 450:1026–1030. doi:10.1038/nature06387
Hessa T, Sharma A, Mariappan M, Eshleman HD, Gutierrez E, Hegde RS.
2011. Protein targeting and degradation are coupled for elimination of
mislocalized proteins. Nature 475:394–397.
doi:10.1038/nature10181
Howe KL, Contreras-Moreira B, De Silva N, Maslen G, Akanni W, Allen J,
Alvarez-Jarreta J, Barba M, Bolser DM, Cambell L, Carbajo M,
Chakiachvili M, Christensen M, Cummins C, Cuzick A, Davis P, Fexova S,
Gall A, George N, Gil L, Gupta P, Hammond-Kosack KE, Haskell E, Hunt SE,
Jaiswal P, Janacek SH, Kersey PJ, Langridge N, Maheswari U, Maurel T,
McDowall MD, Moore B, Muffato M, Naamati G, Naithani S, Olson A,
Papatheodorou I, Patricio M, Paulini M, Pedro H, Perry E, Preece J,
Rosello M, Russell M, Sitnik V, Staines DM, Stein J, Tello-Ruiz MK,
Trevanion SJ, Urban M, Wei S, Ware D, Williams G, Yates AD, Flicek P.
2020. Ensembl Genomes 2020-enabling non-vertebrate genomic
research. Nucleic Acids Res 48:D689–D695.
doi:10.1093/nar/gkz890
Itakura E, Zavodszky E, Shao S, Wohlever ML, Keenan RJ, Hegde RS. 2016.
Ubiquilins Chaperone and Triage Mitochondrial
Membrane Proteins for Degradation. Mol Cell
63:21–33. doi:10.1016/j.molcel.2016.05.020
Jahandideh S, Jaroszewski L, Godzik A. 2014. Improving the chances of
successful protein structure determination with a random forest
classifier. Acta Crystallographica Section D, Biological
Crystallography 70:627–635. doi:10.1107/S1399004713032070
Jenny B. 2020. Color
Oracle: Design for the Color
Impaired.
Joachims T. 2002. Optimizing search engines using clickthrough data.
ACM Press. p. 133. doi:10.1145/775047.775067
Johansson LC, Stauch B, Ishchenko A, Cherezov V. 2017. A bright future
for serial femtosecond crystallography with XFELs.
Trends in Biochemical Sciences 42:749–762.
doi:10.1016/j.tibs.2017.06.007
Kabsch W, Sander C. 1983. Dictionary of protein secondary structure:
Pattern recognition of hydrogen-bonded and geometrical features.
Biopolymers 22:2577–2637. doi:10.1002/bip.360221211
Kajander T, Sachs JN, Goldman A, Regan L. 2009. Electrostatic
interactions of Hsp-organizing protein
tetratricopeptide domains with Hsp70 and
Hsp90: Computational analysis and protein engineering.
J Biol Chem 284:25364–25374. doi:10.1074/jbc.M109.033894
Kalbfleisch T, Cambon A, Wattenberg BW. 2007. A bioinformatics approach
to identifying tail-anchored proteins in the human genome.
Traffic 8:1687–1694. doi:10.1111/j.1600-0854.2007.00661.x
Käll L, Krogh A, Sonnhammer ELL. 2004. A combined transmembrane topology
and signal peptide prediction method. Journal of Molecular
Biology 338:1027–1036. doi:10.1016/j.jmb.2004.03.016
Katoh K, Standley DM. 2013. MAFFT multiple sequence
alignment software version 7: Improvements in performance and usability.
Mol Biol Evol 30:772–780. doi:10.1093/molbev/mst010
Keenan RJ, Freymann DM, Walter P, Stroud RM. 1998. Crystal structure of
the signal sequence binding subunit of the signal recognition particle.
Cell 94:181–191. doi:10.1016/s0092-8674(00)81418-x
Kendall MG. 1938. A new measure of rank correlation. Biometrika
30:81. doi:10.2307/2332226
Kiktev DA, Patterson JC, Müller S, Bariar B, Pan T, Chernoff YO. 2012.
Regulation of chaperone effects on a yeast prion by cochaperone
Sgt2. Mol Cell Biol 32:4960–4970.
doi:10.1128/MCB.00875-12
Kirschke E, Goswami D, Southworth D, Griffin PR, Agard DA. 2014.
Glucocorticoid receptor function regulated by coordinated action of the
Hsp90 and Hsp70 chaperone cycles.
Cell 157:1685–1697. doi:10.1016/j.cell.2014.04.038
Ko HS, Uehara T, Tsuruma K, Nomura Y. 2004. Ubiquilin interacts with
ubiquitylated proteins and proteasome through its ubiquitin-associated
and ubiquitin-like domains. FEBS Lett
566:110–114. doi:10.1016/j.febslet.2004.04.031
Ko K, Banerjee S, Innes J, Taylor D, Ko Z. 2004. The Tic40
translocon components exhibit preferential interactions with different
forms of the Oee1 plastid protein precursor. Funct
Plant Biol 31:285–294. doi:10.1071/FP03195
Köhler RH. 1998. GFP for in vivo imaging of subcellular
structures in plant cells. Trends in Plant Science
3:317–320. doi:10.1016/S1360-1385(98)01276-X
Koonin EV. 1993. A superfamily of ATPases with diverse
functions containing either classical or deviant ATP-binding motif. J Mol Biol
229:1165–1174. doi:10.1006/jmbi.1993.1115
Korepanova A, Gao FP, Hua Y, Qin H, Nakamoto RK, Cross TA. 2005. Cloning
and expression of multiple integral membrane proteins from
Mycobacterium tuberculosis in Escherichia
coli. Protein Science 14:148–158. doi:10.1110/ps.041022305
Kotoshiba S, Kamura T, Hara T, Ishida N, Nakayama KI. 2005. Molecular
dissection of the interaction between P27 and Kip1
ubiquitylation-promoting complex, the ubiquitin ligase that regulates
proteolysis of P27 in G1 phase. J Biol Chem
280:17694–17700. doi:10.1074/jbc.M500866200
Kovacheva S, Bédard J, Patel R, Dudley P, Twell D, Ríos G, Koncz C,
Jarvis P. 2005. In vivo studies on the roles of Tic110,
Tic40 and Hsp93 during chloroplast protein
import. Plant J 41:412–428. doi:10.1111/j.1365-313X.2004.02307.x
Kovalevskiy O, Nicholls RA, Long F, Carlon A, Murshudov GN. 2018.
Overview of refinement procedures within REFMAC5: Utilizing
data from different sources. Acta Crystallogr D Struct Biol
74:215–227. doi:10.1107/S2059798318000979
Kovesi P. 2015. Good
Colour Maps: How to Design
Them.
Krogh A, Larsson B, von Heijne G, Sonnhammer EL. 2001. Predicting
transmembrane protein topology with a hidden Markov model:
Application to complete genomes. J Mol Biol
305:567–580. doi:10.1006/jmbi.2000.4315
Kuhn M. 2008. Building predictive models in R using the
caret package. J of Stat Soft.
Kunze M, Berger J. 2015. The similarity between N-terminal targeting signals for protein import
into different organelles and its evolutionary relevance. Front
Physiol 6:259. doi:10.3389/fphys.2015.00259
Kutay U, Hartmann E, Rapoport TA. 1993. A class of membrane proteins
with a C-terminal anchor. Trends Cell
Biol 3:72–75. doi:10.1016/0962-8924(93)90066-a
Kyte J, Doolittle RF. 1982. A simple method for displaying the
hydropathic character of a protein. J Mol Biol
157:105–132. doi:10.1016/0022-2836(82)90515-0
Lässle M, Blatch GL, Kundra V, Takatori T, Zetter BR. 1997.
Stress-inducible, murine protein mSTI1.
Characterization of binding domains for heat shock proteins
and in vitro phosphorylation by different kinases. J Biol Chem
272:1876–1884. doi:10.1074/jbc.272.3.1876
Lechat P, Hummel L, Rousseau S, Moszer I. 2008. GenoList:
An integrated environment for comparative analysis of microbial genomes.
Nucleic Acids Research 36:D469–474. doi:10.1093/nar/gkm1042
Lee DW, Lee J, Hwang I. 2017. Sorting of nuclear-encoded chloroplast
membrane proteins. Curr Opin Plant Biol
40:1–7. doi:10.1016/j.pbi.2017.06.011
Leipe DD, Wolf YI, Koonin EV, Aravind L. 2002. Classification and
evolution of P-loop GTPases and related
ATPases. J Mol Biol 317:41–72.
doi:10.1006/jmbi.2001.5378
Letunic I, Bork P. 2018. 20 years of the SMART protein
domain annotation resource. Nucleic Acids Res
46:D493–D496. doi:10.1093/nar/gkx922
Letunic I, Doerks T, Bork P. 2015. SMART: Recent updates,
new developments and status in 2015. Nucleic Acids Res
43:D257–260. doi:10.1093/nar/gku949
Levchenko M, Wuttke J-M, Römpler K, Schmidt B, Neifer K, Juris L, Wissel
M, Rehling P, Deckers M. 2016. Cox26 is a novel stoichiometric subunit
of the yeast cytochrome c oxidase. Biochim Biophys Acta
1863:1624–1632. doi:10.1016/j.bbamcr.2016.04.007
Lewinson O, Lee AT, Rees DC. 2008. The funnel approach to the
precrystallization production of membrane proteins. Journal of
Molecular Biology 377:62–73. doi:10.1016/j.jmb.2007.12.059
Leznicki P, High S. 2012. SGTA antagonizes BAG6-mediated protein triage. Proc Natl Acad
Sci U S A 109:19214–19219. doi:10.1073/pnas.1209997109
Leznicki P, Korac-Prlic J, Kliza K, Husnjak K, Nyathi Y, Dikic I, High
S. 2015. Binding of SGTA to Rpn13 selectively
modulates protein quality control. J Cell Sci
128:3187–3196. doi:10.1242/jcs.165209
Li G-W, Oh E, Weissman JS. 2012. The anti-Shine-Dalgarno
sequence drives translational pausing and codon choice in bacteria.
Nature 484:538–541. doi:10.1038/nature10965
Li W, Godzik A. 2006. Cd-hit: A fast program for clustering and
comparing large sets of protein or nucleotide sequences.
Bioinformatics 22:1658–1659. doi:10.1093/bioinformatics/btl158
Li Z, Hartl FU, Bracher A. 2013. Structure and function of
Hip, an attenuator of the Hsp70 chaperone
cycle. Nat Struct Mol Biol 20:929–935. doi:10.1038/nsmb.2608
Liebschner D, Afonine PV, Baker ML, Bunkóczi G, Chen VB, Croll TI,
Hintze B, Hung L-W, Jain S, McCoy AJ, Moriarty NW, Oeffner RD, Poon BK,
Prisant MG, Read RJ, Richardson JS, Richardson DC, Sammito MD, Sobolev
OV, Stockwell DH, Terwilliger TC, Urzhumtsev AG, Videau LL, Williams CJ,
Adams PD. 2019. Macromolecular structure determination using X-rays, neutrons and electrons: Recent
developments in Phenix. Acta Crystallogr D
Struct Biol 75:861–877. doi:10.1107/S2059798319011471
Lim PJ, Danner R, Liang J, Doong H, Harman C, Srinivasan D, Rothenberg
C, Wang H, Ye Y, Fang S, Monteiro MJ. 2009. Ubiquilin and
P97/VCP bind erasin, forming a complex involved in
ERAD. J Cell Biol 187:201–217.
doi:10.1083/jcb.200903024
Lin K-F, Fry MY, Saladi SM, Clemons WM. 2021. Molecular basis of
tail-anchored integral membrane protein recognition by the cochaperone
Sgt2. J Biol Chem 296:100441.
doi:10.1016/j.jbc.2021.100441
Linding R, Jensen LJ, Diella F, Bork P, Gibson TJ, Russell RB. 2003.
Protein disorder prediction: Implications for structural proteomics.
Structure 11:1453–1459.
Liou S-T, Wang C. 2005. Small glutamine-rich tetratricopeptide
repeat-containing protein is composed of three structural units with
distinct functions. Arch Biochem Biophys
435:253–263. doi:10.1016/j.abb.2004.12.020
Liu FH, Wu SJ, Hu SM, Hsiao CD, Wang C. 1999. Specific interaction of
the 70-kDa heat shock cognate protein with
the tetratricopeptide repeats. J Biol Chem
274:34425–34432. doi:10.1074/jbc.274.48.34425
Long P, Samnakay P, Jenner P, Rose S. 2012. A yeast two-hybrid screen
reveals that osteopontin associates with MAP1A and
MAP1B in addition to other proteins linked to microtubule
stability, apoptosis and protein degradation in the human brain. Eur
J Neurosci 36:2733–2742. doi:10.1111/j.1460-9568.2012.08189.x
Lorenz R, Bernhart SH, Höner Zu Siederdissen C, Tafer H, Flamm C,
Stadler PF, Hofacker IL. 2011. ViennaRNA package 2.0.
Algorithms for molecular biology: AMB 6:26.
doi:10.1186/1748-7188-6-26
Lott A, Oroz J, Zweckstetter M. 2020. Molecular basis of the interaction
of Hsp90 with its co-chaperone Hop.
Protein Sci 29:2422–2432. doi:10.1002/pro.3969
Love J, Mancia F, Shapiro L, Punta M, Rost B, Girvin M, Wang D-N, Zhou
M, Hunt JF, Szyperski T, Gouaux E, MacKinnon R, McDermott A, Honig B,
Inouye M, Montelione G, Hendrickson WA. 2010. The New York
Consortium on Membrane Protein Structure
(NYCOMPS): A high-throughput platform for structural
genomics of integral membrane proteins. Journal of Structural and
Functional Genomics 11:191–199. doi:10.1007/s10969-010-9094-7
Lowe ED, Hasan N, Trempe JF, Fonso L, Noble MEM, Endicott JA, Johnson
LN, Brown NR. 2006. Structures of the Dsk2 UBL and
UBA domains and their complex. Acta Crystallogr D Biol
Crystallogr 62:177–188. doi:10.1107/S0907444905037777
Lu AX, Zarin T, Hsu IS, Moses AM. 2019. YeastSpotter:
Accurate and parameter-free web segmentation for microscopy images of
yeast cells. Bioinformatics 35:4525–4527.
doi:10.1093/bioinformatics/btz402
Lundstrom K, Wagner R, Reinhart C, Desmyter A, Cherouati N, Magnin T,
Zeder-Lutz G, Courtot M, Prual C, André N, Hassaine G, Michel H,
Cambillau C, Pattus F. 2006. Structural genomics on membrane proteins:
Comparison of more than 100 GPCRs in 3 expression systems.
Journal of Structural and Functional Genomics
7:77–91. doi:10.1007/s10969-006-9011-2
Luo P, Baldwin RL. 1997. Mechanism of helix induction by
trifluoroethanol: A framework for extrapolating the helix-forming
properties of peptides from trifluoroethanol/water mixtures back to
water. Biochemistry 36:8413–8421. doi:10.1021/bi9707133
Ma P, Varela F, Magoch M, Silva AR, Rosário AL, Brito J, Oliveira TF,
Nogly P, Pessanha M, Stelter M, Kletzin A, Henderson PJF, Archer M.
2013. An efficient strategy for small-scale screening and production of
archaeal membrane transport proteins in Escherichia coli.
PloS One 8:e76913. doi:10.1371/journal.pone.0076913
Machado GM, Oliveira MM, Fernandes L. 2009. A Physiologically-based Model for
Simulation of Color Vision Deficiency.
IEEE Trans Visual Comput Graphics
15:1291–1298. doi:10.1109/TVCG.2009.113
Madhavan V, Bhatt F, Jeffery CJ. 2010. Recombinant expression screening
of P. Aeruginosa bacterial inner membrane proteins. BMC
biotechnology 10:83. doi:10.1186/1472-6750-10-83
Maggiolo AO, Mahajan S, Rees DC, Clemons WM. 2023. Intradimeric
Walker A ATPases: Conserved Features of
A Functionally Diverse Family. J Mol Biol 167965.
doi:10.1016/j.jmb.2023.167965
Manu MS, Ghosh D, Chaudhari BP, Ramasamy S. 2018. Analysis of
tail-anchored protein translocation pathway in plants. Biochem
Biophys Rep 14:161–167. doi:10.1016/j.bbrep.2018.05.001
Mareš J, Strunecký O, Bučinská L, Wiedermannová J. 2019. Evolutionary
Patterns of Thylakoid Architecture in
Cyanobacteria. Front Microbiol
10:277. doi:10.3389/fmicb.2019.00277
Marín I. 2014. The ubiquilin gene family: Evolutionary patterns and
functional insights. BMC Evol Biol 14:63.
doi:10.1186/1471-2148-14-63
Marshall SS, Niesen MJM, Müller A, Tiemann K, Saladi SM, Galimidi RP,
Zhang B, Clemons WM, Miller TF. 2016. A link between integral membrane
protein expression and simulated integration efficiency. Cell
Reports 16:2169–2177. doi:10.1016/j.celrep.2016.07.042
Martínez-Lumbreras S, Krysztofinska EM, Thapaliya A, Spilotros A,
Matak-Vinkovic D, Salvadori E, Roboti P, Nyathi Y, Muench JH, Roessler
MM, Svergun DI, High S, Isaacson RL. 2018. Structural complexity of the
co-chaperone SGTA: A conserved C-terminal region is implicated in dimerization
and substrate quality control. BMC Biol 16:76.
doi:10.1186/s12915-018-0542-3
Martins VR, Graner E, Garcia-Abreu J, de Souza SJ, Mercadante AF, Veiga
SS, Zanata SM, Neto VM, Brentani RR. 1997. Complementary hydropathy
identifies a cellular prion protein receptor. Nat Med
3:1376–1382. doi:10.1038/nm1297-1376
Mateja A, Szlachcic A, Downing ME, Dobosz M, Mariappan M, Hegde RS,
Keenan RJ. 2009. The structural basis of tail-anchored membrane protein
recognition by Get3. Nature
461:361–366. doi:10.1038/nature08319
McGibbon RT, Beauchamp KA, Harrigan MP, Klein C, Swails JM, Hernández
CX, Schwantes CR, Wang L-P, Lane TJ, Pande VS. 2015.
MDTraj: A Modern Open Library for the
Analysis of Molecular Dynamics Trajectories.
Biophys J 109:1528–1532. doi:10.1016/j.bpj.2015.08.015
McMurdie PJ, Holmes S. 2013. Phyloseq: An R package for
reproducible interactive analysis and graphics of microbiome census
data. PLoS One 8:e61217. doi:10.1371/journal.pone.0061217
Merk A, Bartesaghi A, Banerjee S, Falconieri V, Rao P, Davis MI, Pragani
R, Boxer MB, Earl LA, Milne JLS, Subramaniam S. 2016. Breaking
cryo-EM resolution barriers to facilitate drug discovery.
Cell 165:1698–1707. doi:10.1016/j.cell.2016.05.040
Micsonai A, Wien F, Kernya L, Lee Y-H, Goto Y, Réfrégiers M, Kardos J.
2015. Accurate secondary structure prediction and fold recognition for
circular dichroism spectroscopy. Proc Natl Acad Sci U S A
112:E3095–3103. doi:10.1073/pnas.1500851112
Mirzadeh K, Martínez V, Toddo S, Guntur S, Herrgård MJ, Elofsson A,
Nørholm MHH, Daley DO. 2015. Enhanced protein production in escherichia
coli by optimization of cloning scars at the vector-coding sequence
junction. ACS synthetic biology. doi:10.1021/acssynbio.5b00033
Mistry J, Chuguransky S, Williams L, Qureshi M, Salazar GA, Sonnhammer
ELL, Tosatto SCE, Paladin L, Raj S, Richardson LJ, Finn RD, Bateman A.
2021. Pfam: The protein families database in 2021.
Nucleic Acids Res 49:D412–D419. doi:10.1093/nar/gkaa913
Mock J-Y, Chartron JW, Zaslaver M, Xu Y, Ye Y, Clemons WM. 2015. Bag6
complex contains a minimal tail-anchor-targeting module and a mock
BAG domain. Proc Natl Acad Sci U S A
112:106–111. doi:10.1073/pnas.1402745112
Moore KR, Magnabosco C, Momper L, Gold DA, Bosak T, Fournier GP. 2019.
An Expanded Ribosomal Phylogeny of Cyanobacteria
Supports a Deep Placement of Plastids.
Front Microbiol 10:1612. doi:10.3389/fmicb.2019.01612
Moreland K. 2016. Why we use bad color maps and what you can do about
it. Electronic Imaging 2016:1–6. doi:10.2352/ISSN.2470-1173.2016.16.HVEI-133
Moroney N, Fairchild MD, Hunt RW, Li C, Luo MR, Newman T. 2002. The
CIECAM02 color appearance modelColor and Imaging
Conference. Society for Imaging Science and
Technology. pp. 23–27.
Nannenga BL, Gonen T. 2016. MicroED opens a new era for
biological structure determination. Current Opinion in Structural
Biology 40:128–135. doi:10.1016/j.sbi.2016.09.007
Nelson GM, Huffman H, Smith DF. 2003. Comparison of the carboxy-terminal
DP-repeat region in the co-chaperones
Hop and Hip. Cell Stress Chaperones
8:125–133. doi:10.1379/1466-1268(2003)008<0125:cotcdr>2.0.co;2
Nelson KE, Clayton RA, Gill SR, Gwinn ML, Dodson RJ, Haft DH, Hickey EK,
Peterson JD, Nelson WC, Ketchum KA, McDonald L, Utterback TR, Malek JA,
Linher KD, Garrett MM, Stewart AM, Cotton MD, Pratt MS, Phillips CA,
Richardson D, Heidelberg J, Sutton GG, Fleischmann RD, Eisen JA, White
O, Salzberg SL, Smith HO, Venter JC, Fraser CM. 1999. Evidence for
lateral gene transfer between Archaea and bacteria from
genome sequence of Thermotoga maritima. Nature
399:323–329. doi:10.1038/20601
Neuwirth E. 2014. RColorBrewer:
ColorBrewer palettes.
Nielsen H. 2017. Predicting Secretory Proteins with
SignalP. Methods Mol Biol
1611:59–73. doi:10.1007/978-1-4939-7015-5_6
Niesen MJM, Marshall SS, Miller TF, Clemons WM. 2017. Improving membrane
protein expression by optimizing integration efficiency. The Journal
of Biological Chemistry. doi:10.1074/jbc.M117.813469
Nørholm MHH, Light S, Virkki MTI, Elofsson A, von Heijne G, Daley DO.
2012. Manipulating the genetic code for membrane protein production:
What have we learnt so far? Biochimica et biophysica acta
1818:1091–1096. doi:10.1016/j.bbamem.2011.08.018
Nørholm MHH, Toddo S, Virkki MTI, Light S, von Heijne G, Daley DO. 2013.
Improved production of membrane proteins in Escherichia
coli by selective codon substitutions. FEBS letters
587:2352–2358. doi:10.1016/j.febslet.2013.05.063
Nuñez JR, Anderton CR, Renslow RS. 2018. Optimizing colormaps with
consideration for color vision deficiency to enable accurate
interpretation of scientific data. PLoS ONE
13:e0199239. doi:10.1371/journal.pone.0199239
Oborník M. 2019. Endosymbiotic Evolution of
Algae, Secondary Heterotrophy and
Parasitism. Biomolecules 9:266.
doi:10.3390/biom9070266
Okabe M, Ito K. 2002. How to make figures and presentations that are
friendly to color blind people. http://jfly.iam.u-tokyo.ac.jp/html/color_blind/
Okreglak V, Walter P. 2014. The conserved AAA-ATPase Msp1
confers organelle specificity to tail-anchored proteins. Proc Natl
Acad Sci U S A 111:8019–8024. doi:10.1073/pnas.1405755111
Onuoha SC, Coulstock ET, Grossmann JG, Jackson SE. 2008. Structural
studies on the co-chaperone Hop and its complexes with
Hsp90. J Mol Biol 379:732–744.
doi:10.1016/j.jmb.2008.02.013
Pace CN, Scholtz JM. 1998. A helix propensity scale based on
experimental studies of peptides and proteins. Biophys J
75:422–427. doi:10.1016/s0006-3495(98)77529-0
Pattengale ND, Alipour M, Bininda-Emonds ORP, Moret BME, Stamatakis A.
2010. How many bootstrap replicates are necessary? J Comput
Biol 17:337–354. doi:10.1089/cmb.2009.0179
Peden JF. 2000. Analysis of codon usage (PhD thesis). University
of Nottingham.
Pei J, Kim B-H, Grishin NV. 2008. PROMALS3D: A tool for
multiple protein sequence and structure alignments. Nucleic Acids
Res 36:2295–2300. doi:10.1093/nar/gkn072
Petrovskaya LE, Shulga AA, Bocharova OV, Ermolyuk YS, Kryukova EA,
Chupin VV, Blommers MJJ, Arseniev AS, Kirpichnikov MP. 2010. Expression
of G-protein coupled receptors in
Escherichia coli for structural studies. Biochemistry
(Moscow) 75:881–891.
Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris
JH, Ferrin TE. 2021. UCSF
ChimeraX : Structure visualization for
researchers, educators, and developers. Protein Science
30:70–82. doi:10.1002/pro.3943
Pieper U, Schlessinger A, Kloppmann E, Chang GA, Chou JJ, Dumont ME, Fox
BG, Fromme P, Hendrickson WA, Malkowski MG, Rees DC, Stokes DL, Stowell
MHB, Wiener MC, Rost B, Stroud RM, Stevens RC, Sali A. 2013.
Coordinating the impact of structural genomics on the human a-helical
transmembrane proteome. Nat Struct Mol Biol
20:135–138. doi:10.1038/nsmb.2508
Pierce BG, Wiehe K, Hwang H, Kim B-H, Vreven T, Weng Z. 2014.
ZDOCK server: Interactive docking prediction of
protein-protein complexes and symmetric multimers.
Bioinformatics 30:1771–1773. doi:10.1093/bioinformatics/btu097
Prapapanich V, Chen S, Smith DF. 1998. Mutation of Hip’s
carboxy-terminal region inhibits a transitional stage of progesterone
receptor assembly. Mol Cell Biol 18:944–952.
doi:10.1128/MCB.18.2.944
Prapapanich V, Chen S, Toran EJ, Rimerman RA, Smith DF. 1996. Mutational
analysis of the Hsp70-interacting protein Hip. Mol Cell
Biol 16:6200–6207. doi:10.1128/MCB.16.11.6200
Price WN, Chen Y, Handelman SK, Neely H, Manor P, Karlin R, Nair R, Liu
J, Baran M, Everett J, Tong SN, Forouhar F, Swaminathan SS, Acton T,
Xiao R, Luft JR, Lauricella A, DeTitta GT, Rost B, Montelione GT, Hunt
JF. 2009. Understanding the physical properties that control protein
crystallization by analysis of large-scale experimental data. Nature
Biotechnology 27:51–57. doi:10.1038/nbt.1514
Prodromou C. 2016. Mechanisms of Hsp90 regulation.
Biochem J 473:2439–2452. doi:10.1042/BCJ20160005
Psakis G, Nitschkowski S, Holz C, Kress D, Maestre-Reyna M, Polaczek J,
Illing G, Essen L-O. 2007. Expression screening of integral membrane
proteins from Helicobacter pylori 26695. Protein
Science 16:2667–2676. doi:10.1110/ps.073104707
Punta M, Love J, Handelman S, Hunt JF, Shapiro L, Hendrickson WA, Rost
B. 2009. Structural genomics target selection for the New
York consortium on membrane protein structure. Journal of
Structural and Functional Genomics 10:255–268.
doi:10.1007/s10969-009-9071-1
R Core Team. 2015. R: A
Language and Environment for Statistical
Computing.
Rabu C, Schmid V, Schwappach B, High S. 2009. Biogenesis of
tail-anchored proteins: The beginning for the end? J Cell Sci
122:3605–3612. doi:10.1242/jcs.041210
Rao M, Okreglak V, Chio US, Cho H, Walter P, Shan S-O. 2016. Multiple
selection filters ensure accurate tail-anchored membrane protein
targeting. Elife 5:e21301. doi:10.7554/eLife.21301
Raymond J, Blankenship RE. 2003. Horizontal gene transfer in eukaryotic
algal evolution. Proc Natl Acad Sci U S A
100:7419–7420. doi:10.1073/pnas.1533212100
Reidy M, Kumar S, Anderson DE, Masison DC. 2018. Dual Roles
for Yeast Sti1/Hop in Regulating
the Hsp90 Chaperone Cycle. Genetics
209:1139–1154. doi:10.1534/genetics.118.301178
Reis AC, Salis HM. 2020. An Automated Model Test System for
Systematic Development and Improvement of
Gene Expression Models. ACS Synth Biol
9:3145–3156. doi:10.1021/acssynbio.0c00394
Reißer S, Strandberg E, Steinbrecher T, Ulrich AS. 2014. 3D
hydrophobic moment vectors as a tool to characterize the surface
polarity of amphiphilic peptides. Biophys J
106:2385–2394. doi:10.1016/j.bpj.2014.04.020
Revell LJ. 2012. Phytools: An R package for phylogenetic
comparative biology (and other things). Methods in ecology and
evolution 217–223.
Revolution Analytics, Weston S. 2015a. doMC: Foreach Parallel Adaptor for
’parallel’.
Revolution Analytics, Weston S. 2015b. doParallel: Foreach Parallel Adaptor
for the ’parallel’ Package.
Revolution Analytics, Weston S. 2015c. Foreach: Provides
Foreach Looping Construct for R.
Revolution Analytics, Weston S. 2015d. Iterators:
Provides Iterator Construct for R.
Richardson JS. 2000. Early ribbon drawings of proteins. Nature
Structural Biology 7:624–625. doi:10.1038/77912
Ries F, Herkt C, Willmund F. 2020. Co-Translational Protein
Folding and Sorting in Chloroplasts.
Plants (Basel) 9:214. doi:10.3390/plants9020214
Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez J-C, Müller
M. 2011. pROC: An open-source package for
R and S+ to analyze and compare
ROC curves. BMC bioinformatics
12:77. doi:10.1186/1471-2105-12-77
Rodrigo-Brenni MC, Gutierrez E, Hegde RS. 2014. Cytosolic quality
control of mislocalized proteins requires RNF126
recruitment to Bag6. Mol Cell
55:227–237. doi:10.1016/j.molcel.2014.05.025
Rogowitz BE, Kalvin AD. 2001. The "Which Blair project": A
quick visual method for evaluating perceptual color mapsProceedings
Visualization, 2001. VIS ’01. Presented at the
VIS 2001. Visualization 2001. San Diego,
CA, USA: IEEE. pp. 183–556. doi:10.1109/VISUAL.2001.964510
Röhl A, Rohrberg J, Buchner J. 2013. The chaperone Hsp90:
Changing partners for demanding clients. Trends Biochem Sci
38:253–262. doi:10.1016/j.tibs.2013.02.003
Röhl A, Wengler D, Madl T, Lagleder S, Tippel F, Herrmann M, Hendrix J,
Richter K, Hack G, Schmid AB, Kessler H, Lamb DC, Buchner J. 2015. Hsp90
regulates the dynamics of its cochaperone Sti1 and the
transfer of Hsp70 between modules. Nat Commun
6:6655. doi:10.1038/ncomms7655
Rome ME, Chio US, Rao M, Gristick H, Shan S. 2014. Differential
gradients of interaction affinities drive efficient targeting and
recycling in the GET pathway. Proc Natl Acad Sci U S
A 111:E4929–4935. doi:10.1073/pnas.1411284111
Rome ME, Rao M, Clemons WM, Shan S. 2013. Precise timing of
ATPase activation drives targeting of tail-anchored
proteins. Proc Natl Acad Sci U S A
110:7666–7671. doi:10.1073/pnas.1222054110
Roseman MA. 1988. Hydrophilicity of polar amino acid side-chains is
markedly reduced by flanking peptide bonds. J Mol Biol
200:513–522. doi:10.1016/0022-2836(88)90540-2
Rosen BP, Weigel U, Karkaria C, Gangola P. 1988. Molecular
characterization of a unique anion pump: The ArsA protein
is an arsenite(antimonate)-stimulated ATPase. Prog Clin
Biol Res 273:105–112.
RStudio Team. 2015. RStudio: Integrated Development
Environment for R.
Russell RB, Barton GJ. 1992. Multiple protein sequence alignment from
tertiary structure comparison: Assignment of global and residue
confidence levels. Proteins 14:309–323. doi:10.1002/prot.340140216
Saeki Y, Sone T, Toh-e A, Yokosawa H. 2002. Identification of
ubiquitin-like protein-binding subunits of the 26S
proteasome. Biochem Biophys Res Commun
296:813–819. doi:10.1016/s0006-291x(02)02002-8
Saier MH, Reddy VS, Tsu BV, Ahmed MS, Li C, Moreno-Hagelsieb G. 2016.
The transporter classification database (TCDB): Recent
advances. Nucleic Acids Research 44:D372–379.
doi:10.1093/nar/gkv1103
Saladi SM, Maggiolo AO, Radford K, Clemons WM. 2020. Structural
biologists, let’s mind our colors (preprint). Biochemistry.
doi:10.1101/2020.09.22.308593
Sarkar CA, Dodevski I, Kenig M, Dudli S, Mohr A, Hermans E, Plückthun A.
2008. Directed evolution of a G protein-coupled receptor
for expression, stability, and binding selectivity. Proceedings of
the National Academy of Sciences of the United States of America
105:14808–14813. doi:10.1073/pnas.0803103105
Schägger H. 2006. Tricine-SDS-PAGE. Nat Protoc
1:16–22. doi:10.1038/nprot.2006.4
Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H,
Hartl FU, Moarefi I. 2000. Structure of TPR domain-peptide
complexes: Critical elements in the assembly of the
Hsp70-Hsp90 multichaperone machine. Cell
101:199–210. doi:10.1016/S0092-8674(00)80830-2
Schiffer M, Edmundson AB. 1967. Use of helical wheels to represent the
structures of proteins and to identify segments with helical potential.
Biophys J 7:121–135. doi:10.1016/S0006-3495(67)86579-2
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch
T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ,
Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: An
open-source platform for biological-image analysis. Nat Methods
9:676–682. doi:10.1038/nmeth.2019
Schlinkmann KM, Honegger A, Türeci E, Robison KE, Lipovšek D, Plückthun
A. 2012. Critical features for biosynthesis, stability, and
functionality of a G protein-coupled receptor uncovered by
all-versus-all mutations. Proceedings of the National Academy of
Sciences of the United States of America
109:9810–9815. doi:10.1073/pnas.1202107109
Schloerke B, Crowley J, Cook D, Briatte F, Marbach M, Thoen E, Elberg A,
Larmarange J. 2016. GGally:
Extension to ’Ggplot2’.
Schmid AB, Lagleder S, Gräwert MA, Röhl A, Hagn F, Wandinger SK, Cox MB,
Demmer O, Richter K, Groll M, Kessler H, Buchner J. 2012. The
architecture of functional modules in the Hsp90
co-chaperone Sti1/Hop. EMBO J
31:1506–1517. doi:10.1038/emboj.2011.472
Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to
ImageJ: 25 years of image analysis. Nat Methods
9:671–675. doi:10.1038/nmeth.2089
Schopf FH, Biebl MM, Buchner J. 2017. The HSP90 chaperone
machinery. Nat Rev Mol Cell Biol 18:345–360.
doi:10.1038/nrm.2017.20
Schuldiner M, Metz J, Schmid V, Denic V, Rakwalska M, Schmitt HD,
Schwappach B, Weissman JS. 2008. The GET complex mediates
insertion of tail-anchored proteins into the ER membrane.
Cell 134:634–645. doi:10.1016/j.cell.2008.06.025
Schuler GD, Epstein JA, Ohkawa H, Kans JA. 1996. Entrez: Molecular
biology database and retrieval system. Methods in Enzymology
266:141–162.
Şentürk M, Lin G, Zuo Z, Mao D, Watson E, Mikos AG, Bellen HJ. 2019.
Ubiquilins regulate autophagic flux through mTOR signalling and lysosomal acidification.
Nat Cell Biol 21:384–396. doi:10.1038/s41556-019-0281-x
Seppälä S, Slusky JS, Lloris-Garcerá P, Rapp M, von Heijne G. 2010.
Control of membrane protein topology by a single C-terminal residue. Science (New York,
NY) 328:1698–1700. doi:10.1126/science.1188950
Serres MH, Kerr ARW, McCormack TJ, Riley M. 2009. Evolution by leaps:
Gene duplication in bacteria. Biol Direct
4:46. doi:10.1186/1745-6150-4-46
Shan S-O. 2019. Guiding tail-anchored membrane proteins to the
endoplasmic reticulum in a chaperone cascade. J Biol Chem
294:16577–16586. doi:10.1074/jbc.REV119.006197
Shao S, Hegde RS. 2011a. A calmodulin-dependent translocation pathway
for small secretory proteins. Cell
147:1576–1588. doi:10.1016/j.cell.2011.11.048
Shao S, Hegde RS. 2011b. Membrane protein insertion at the endoplasmic
reticulum. Annu Rev Cell Dev Biol 27:25–56.
doi:10.1146/annurev-cellbio-092910-154125
Shao S, Rodrigo-Brenni MC, Kivlen MH, Hegde RS. 2017. Mechanistic basis
for a molecular triage reaction. Science
355:298–302. doi:10.1126/science.aah6130
Shi T, Falkowski PG. 2008. Genome evolution in cyanobacteria: The stable
core and the variable shell. Proc Natl Acad Sci U S A
105:2510–2515. doi:10.1073/pnas.0711165105
Shindyalov IN, Bourne PE. 1998. Protein structure alignment by
incremental combinatorial extension (CE) of the optimal
path. Protein Eng 11:739–747. doi:10.1093/protein/11.9.739
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. 2015.
BUSCO: Assessing genome assembly and annotation
completeness with single-copy orthologs. Bioinformatics
31:3210–3212. doi:10.1093/bioinformatics/btv351
Simon AC, Simpson PJ, Goldstone RM, Krysztofinska EM, Murray JW, High S,
Isaacson RL. 2013. Structure of the Sgt2/Get5
complex provides insights into GET-mediated
targeting of tail-anchored membrane proteins. Proc Natl Acad Sci U S
A 110:1327–1332. doi:10.1073/pnas.1207518110
Sjores S. 2020. The
following colour schemes have now been implemented.
Slabinski L, Jaroszewski L, Rodrigues APC, Rychlewski L, Wilson IA,
Lesley SA, Godzik A. 2007a. The challenge of protein structure
determination–lessons from structural genomics. Protein Science: A
Publication of the Protein Society 16:2472–2482.
doi:10.1110/ps.073037907
Slabinski L, Jaroszewski L, Rychlewski L, Wilson IA, Lesley SA, Godzik
A. 2007b. XtalPred: A web server for prediction of protein
crystallizability. Bioinformatics (Oxford, England)
23:3403–3405. doi:10.1093/bioinformatics/btm477
Smith NJ, Futrell R, Walt SVD, Mansencal T, TFiFiE, Betts E, Cgohlke,
Dieterle B. 2018. Njsmith/Colorspacious V1.1.2. doi:10.5281/ZENODO.1214904
Smith N, van der Walt S. 2015. A Better
Default Colormap for Matplotlib.
Spatzal T, Aksoyoglu M, Zhang L, Andrade SLA, Schleicher E, Weber S,
Rees DC, Einsle O. 2011. Evidence for Interstitial Carbon
in Nitrogenase FeMo Cofactor. Science
334:940–940. doi:10.1126/science.1214025
Srivastava R, Zalisko BE, Keenan RJ, Howell SH. 2017. The GET
System Inserts the Tail-Anchored Protein,
SYP72, into Endoplasmic Reticulum Membranes.
Plant Physiol 173:1137–1145. doi:10.1104/pp.16.00928
Stamatakis A. 2014. RAxML version 8: A tool for
phylogenetic analysis and post-analysis of large phylogenies.
Bioinformatics 30:1312–1313. doi:10.1093/bioinformatics/btu033
Stefanovic S, Hegde RS. 2007. Identification of a targeting factor for
posttranslational membrane protein insertion into the ER.
Cell 128:1147–1159. doi:10.1016/j.cell.2007.01.036
Stefer S, Reitz S, Wang F, Wild K, Pang Y-Y, Schwarz D, Bomke J, Hein C,
Löhr F, Bernhard F, Denic V, Dötsch V, Sinning I. 2011. Structural basis
for tail-anchored membrane protein biogenesis by the Get3-receptor complex. Science
333:758–762. doi:10.1126/science.1207125
Steinegger M, Söding J. 2018. Clustering huge protein sequence sets in
linear time. Nat Commun 9:2542. doi:10.1038/s41467-018-04964-5
Stringer C, Wang T, Michaelos M, Pachitariu M. 2021. Cellpose: A
generalist algorithm for cellular segmentation. Nat Methods
18:100–106. doi:10.1038/s41592-020-01018-x
Studier FW. 2005. Protein production by auto-induction in high density
shaking cultures. Protein Expr Purif
41:207–234. doi:10.1016/j.pep.2005.01.016
Subudhi I, Shorter J. 2018. Ubiquilin 2: Shuttling Clients
Out of Phase? Mol Cell
69:919–921. doi:10.1016/j.molcel.2018.02.030
Suloway CJM, Chartron JW, Zaslaver M, Clemons WM. 2009. Model for
eukaryotic tail-anchored protein binding based on the structure of
Get3. Proc Natl Acad Sci U S A
106:14849–14854. doi:10.1073/pnas.0907522106
Suloway CJM, Rome ME, Clemons WM. 2012. Tail-anchor targeting by a
Get3 tetramer: The structure of an archaeal homologue.
EMBO J 31:707–719. doi:10.1038/emboj.2011.433
Surade S, Klein M, Stolt-Bergner PC, Muenke C, Roy A, Michel H. 2006.
Comparative analysis and "expression space" coverage of the production
of prokaryotic membrane proteins for structural genomics. Protein
Science 15:2178–2189. doi:10.1110/ps.062312706
Swets JA, Dawes RM, Monahan J. 2000. Better decisions through science.
Sci Am 283:82–87. doi:10.1038/scientificamerican1000-82
Szakonyi G, Leng D, Ma P, Bettaney KE, Saidijam M, Ward A, Zibaei S,
Gardiner AT, Cogdell RJ, Butaye P, Kolsto A-B, O’reilly J, Hope RJ,
Rutherford NG, Hoyle CJ, Henderson PJF. 2007. A genomic strategy for
cloning, expressing and purifying efflux proteins of the major
facilitator superfamily. The Journal of Antimicrobial
Chemotherapy 59:1265–1270. doi:10.1093/jac/dkm036
Tange O. 2011. GNU parallel - the command-line power tool.
;login: The USENIX Magazine 36:42–47. doi:10.5281/zenodo.16303
Ten Eyck LF. 1973. Crystallographic fast Fourier
transforms. Acta Cryst A 29:183–191. doi:10.1107/S0567739473000458
The UniProt Consortium. 2017. UniProt: The universal
protein knowledgebase. Nucleic Acids Res
45:D158–D169. doi:10.1093/nar/gkw1099
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The
CLUSTAL_X windows interface: Flexible
strategies for multiple sequence alignment aided by quality analysis
tools. Nucleic Acids Res 25:4876–4882. doi:10.1093/nar/25.24.4876
Tidow H, Nissen P. 2013. Structural diversity of calmodulin binding to
its target sites. FEBS J 280:5551–5565. doi:10.1111/febs.12296
Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD,
Ketchum KA, Klenk HP, Gill S, Dougherty BA, Nelson K, Quackenbush J,
Zhou L, Kirkness EF, Peterson S, Loftus B, Richardson D, Dodson R,
Khalak HG, Glodek A, McKenney K, Fitzegerald LM, Lee N, Adams MD, Hickey
EK, Berg DE, Gocayne JD, Utterback TR, Peterson JD, Kelley JM, Cotton
MD, Weidman JM, Fujii C, Bowman C, Watthey L, Wallin E, Hayes WS,
Borodovsky M, Karp PD, Smith HO, Fraser CM, Venter JC. 1997. The
complete genome sequence of the gastric pathogen
Helicobacter pylori. Nature
388:539–547. doi:10.1038/41483
Towns J, Cockerill T, Dahan M, Foster I, Gaither K, Grimshaw A,
Hazlewood V, Lathrop S, Lifka D, Peterson GD, Roskies R, Scott JR,
Wilkens-Diehr N. 2014. XSEDE: Accelerating
scientific discovery. Computing in Science & Engineering
16:62–74. doi:10.1109/MCSE.2014.80
Trempe J-F, Šašková KG, Sivá M, Ratcliffe CDH, Veverka V, Hoegl A,
Ménade M, Feng X, Shenker S, Svoboda M, Kožíšek M, Konvalinka J, Gehring
K. 2016. Structural studies of the yeast DNA
damage-inducible protein Ddi1 reveal domain architecture of
this eukaryotic protein family. Sci Rep
6:33671. doi:10.1038/srep33671
Trotta AP, Need EF, Selth LA, Chopra S, Pinnock CB, Leach DA, Coetzee
GA, Butler LM, Tilley WD, Buchanan G. 2013. Knockdown of the cochaperone
SGTA results in the suppression of androgen and
PI3K/Akt signaling and inhibition of prostate
cancer cell proliferation. Int J Cancer
133:2812–2823. doi:10.1002/ijc.28310
Tsirigos KD, Peters C, Shu N, Käll L, Elofsson A. 2015. The
TOPCONS web server for consensus prediction of membrane
protein topology and signal peptides. Nucleic Acids Res
43:W401–407. doi:10.1093/nar/gkv485
Tsochantaridis I, Joachims T, Hofmann T, Altun Y. 2005. Large margin
methods for structured and interdependent output variables. Journal
of Machine Learning Research 6:1453–1484.
UniProt Consortium. 2021. UniProt: The universal protein
knowledgebase in 2021. Nucleic Acids Res
49:D480–D489. doi:10.1093/nar/gkaa1100
UniProt Consortium. 2012. Reorganizing the protein space at the
Universal Protein Resource (UniProt).
Nucleic Acids Res 40:D71–75. doi:10.1093/nar/gkr981
Van den Berg B, Clemons WM, Collinson I, Modis Y, Hartmann E, Harrison
SC, Rapoport TA. 2004. X-ray structure of a protein-conducting channel.
Nature 427:36–44. doi:10.1038/nature02218
van der Walt S, Colbert SC, Varoquaux G. 2011. The NumPy
array: A structure for efficient numerical computation.
Computing in Science & Engineering
13:22–30. doi:10.1109/MCSE.2011.37
Van Lehn RC, Zhang B, Miller TF. 2015. Regulation of multispanning
membrane protein topology via post-translational annealing.
Elife 4. doi:10.7554/eLife.08697
Van Rossum G, Drake Jr FL. 1995. Python reference manual. Centrum
voor Wiskunde en Informatica Amsterdam.
Vida TA, Emr SD. 1995. A new vital stain for visualizing vacuolar
membrane dynamics and endocytosis in yeast. J Cell Biol
128:779–792. doi:10.1083/jcb.128.5.779
Vilardi F, Lorenz H, Dobberstein B. 2011. WRB is the
receptor for TRC40/Asna1-mediated insertion of tail-anchored proteins
into the ER membrane. J Cell Sci
124:1301–1307. doi:10.1242/jcs.084277
Voorhees RM, Hegde RS. 2015. Structures of the scanning and engaged
states of the mammalian SRP-ribosome
complex. Elife 4:e07975. doi:10.7554/eLife.07975
Wade SL, Auble DT. 2010. The Rad23 ubiquitin receptor, the
proteasome and functional specificity in transcriptional control.
Transcription 1:22–26. doi:10.4161/trns.1.1.12201
Wagner S, Klepsch MM, Schlegel S, Appel A, Draheim R, Tarry M, Högbom M,
van Wijk KJ, Slotboom DJ, Persson JO, de Gier J-W. 2008. Tuning
Escherichia coli for membrane protein overexpression.
Proceedings of the National Academy of Sciences of the United States
of America 105:14371–14376. doi:10.1073/pnas.0804090105
Waheed AA, MacDonald S, Khan M, Mounts M, Swiderski M, Xu Y, Ye Y, Freed
EO. 2016. The Vpu-interacting Protein SGTA
Regulates Expression of a Non-glycosylated
Tetherin Species. Sci Rep 6:24934.
doi:10.1038/srep24934
Wallner B, Elofsson A. 2006. Identification of correct regions in
protein models using structural, alignment, and consensus information.
Protein Sci 15:900–913. doi:10.1110/ps.051799606
Wang F, Brown EC, Mak G, Zhuang J, Denic V. 2010. A chaperone cascade
sorts proteins for posttranslational membrane insertion into the
endoplasmic reticulum. Mol Cell 40:159–171.
doi:10.1016/j.molcel.2010.08.038
Wang F, Whynot A, Tung M, Denic V. 2011. The mechanism of tail-anchored
protein insertion into the ER membrane. Mol Cell
43:738–750. doi:10.1016/j.molcel.2011.07.020
Wang H, Feng L, Zhang Z, Webb GI, Lin D, Song J. 2016. Crysalis: An
integrated server for computational analysis and design of protein
crystallization. Scientific Reports 6:21383.
doi:10.1038/srep21383
Wang H, Zhang Q, Zhu D. 2003. hSGT interacts
with the N-terminal region of myostatin.
Biochem Biophys Res Commun 311:877–883. doi:10.1016/j.bbrc.2003.10.080
Wang L-G, Lam TT-Y, Xu S, Dai Z, Zhou L, Feng T, Guo P, Dunn CW, Jones
BR, Bradley T, Zhu H, Guan Y, Jiang Y, Yu G. 2020. Treeio: An R
Package for Phylogenetic Tree Input and
Output with Richly Annotated and
Associated Data. Mol Biol Evol
37:599–603. doi:10.1093/molbev/msz240
Ward N, Moreno-Hagelsieb G. 2014. Quickly finding orthologs as
reciprocal best hits with BLAT, LAST, and
UBLAST: How much do we miss? PLoS One
9:e101850. doi:10.1371/journal.pone.0101850
Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. 2009. Jalview
Version 2–a multiple sequence alignment editor and analysis
workbench. Bioinformatics 25:1189–1191. doi:10.1093/bioinformatics/btp033
Watkins JF, Sung P, Prakash L, Prakash S. 1993. The
Saccharomyces cerevisiae DNA repair gene
RAD23 encodes a nuclear protein containing a ubiquitin-like
domain required for biological function. Mol Cell Biol
13:7757–7765. doi:10.1128/mcb.13.12.7757-7765.1993
Wattenberg B, Lithgow T. 2001. Targeting of C-terminal (tail)-anchored proteins: Understanding
how cytoplasmic activities are anchored to intracellular membranes.
Traffic 2:66–71. doi:10.1034/j.1600-0854.2001.20108.x
Webb B, Sali A. 2016. Comparative Protein Structure Modeling Using
MODELLER. Curr Protoc Bioinformatics
54:5.6.1–5.6.37. doi:10.1002/cpbi.3
Weihs C, Ligges U, Luebke K, Raabe N. 2005. klaR analyzing german business cycles In:
Baier D, Decker R, Schmidt-Thieme L, editors. Data Analysis and Decision
Support. Berlin/Heidelberg: Springer-Verlag.
pp. 335–343.
Weill U, Yofe I, Sass E, Stynen B, Davidi D, Natarajan J, Ben-Menachem
R, Avihou Z, Goldman O, Harpaz N, Chuartzman S, Kniazev K, Knoblach B,
Laborenz J, Boos F, Kowarzyk J, Ben-Dor S, Zalckvar E, Herrmann JM,
Rachubinski RA, Pines O, Rapaport D, Michnick SW, Levy ED, Schuldiner M.
2018. Genome-wide SWAp-Tag yeast libraries for proteome
exploration. Nat Methods 15:617–622. doi:10.1038/s41592-018-0044-9
Weinert K. 2014. Datamart:
Unified access to your data sources.
Weisman CM, Murray AW, Eddy SR. 2020. Many, but not all,
lineage-specific genes can be explained by homology detection failure.
PLoS Biol 18:e3000862. doi:10.1371/journal.pbio.3000862
Wickham H. n.d. Multidplyr:
Partitioned data frames for ’dplyr’.
Wickham H. 2016. Ggplot2:
Elegant graphics for data analysis.
Springer-Verlag New York.
Wickham H. 2015. Scales:
Scale functions for visualization.
Wickham H. 2011. The split-apply-combine strategy for data analysis.
Journal of Statistical Software 40:1–29.
Wickham H, Averick M, Bryan J, Chang W, McGowan LD, François R,
Grolemund G, Hayes A, Henry L, Hester J, others. 2019. Welcome to the
tidyverse. Journal of open source software
4:1686.
Wickham H, Danenberg P, Eugster M. 2015. Roxygen2: In-source documentation for R.
Wickham H, Francois R. 2015. Dplyr: A
grammar of data manipulation.
Wilke CO. 2015. Cowplot:
Streamlined plot theme and plot annotations for
’Ggplot2’.
Wimley WC, Creamer TP, White SH. 1996. Solvation energies of amino acid
side chains and backbone in a family of host-guest pentapeptides.
Biochemistry 35:5109–5124. doi:10.1021/bi9600153
Winkler K-HA, Norman ML. 1986. Munacolor: Understanding high-resolution
gas dynamical simulations through color graphics. Astrophysical
radiation hydrodynamics 223–243.
Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan
RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN,
Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. 2011.
Overview of the CCP4 suite and current developments.
Acta Crystallogr D Biol Crystallogr
67:235–242. doi:10.1107/S0907444910045749
Winnefeld M, Grewenig A, Schnölzer M, Spring H, Knoch TA, Gan EC,
Rommelaere J, Cziepluch C. 2006. Human SGT interacts with
Bag-6/Bat-3/Scythe and cells with
reduced levels of either protein display persistence of few misaligned
chromosomes and mitotic arrest. Exp Cell Res
312:2500–2514. doi:10.1016/j.yexcr.2006.04.020
Wunderley L, Leznicki P, Payapilly A, High S. 2014. SGTA
regulates the cytosolic quality control of hydrophobic substrates. J
Cell Sci 127:4728–4739. doi:10.1242/jcs.155648
Xie Y. 2014. Knitr:
A comprehensive tool for reproducible research in
R In: Stodden V, Leisch F, Peng RD, editors.
Implementing Reproducible Computational Research. Chapman and
Hall/CRC.
Xing S, Mehlhorn DG, Wallmeroth N, Asseck LY, Kar R, Voss A, Denninger
P, Schmidt VAF, Schwarzländer M, Stierhof Y-D, Grossmann G, Grefen C.
2017. Loss of GET pathway orthologs in
Arabidopsis thaliana causes root hair growth defects and
affects SNARE abundance. Proc Natl Acad Sci U S A
114:E1544–E1553. doi:10.1073/pnas.1619525114
Xu D, Zhang Y. 2012. Ab initio protein structure assembly using
continuous structure fragments and optimized knowledge-based force
field. Proteins 80:1715–1735. doi:10.1002/prot.24065
Xu Y, Cai M, Yang Y, Huang L, Ye Y. 2012. SGTA recognizes a
noncanonical ubiquitin-like domain in the Bag6-Ubl4A-Trc35
complex to promote endoplasmic reticulum-associated degradation.
Cell Rep 2:1633–1644. doi:10.1016/j.celrep.2012.11.010
Xu Y, Liu Y, Lee J, Ye Y. 2013. A ubiquitin-like domain recruits an
oligomeric chaperone to a retrotranslocation complex in endoplasmic
reticulum-associated degradation. J Biol Chem
288:18068–18076. doi:10.1074/jbc.M112.449199
Yamamoto Y, Sakisaka T. 2012. Molecular machinery for insertion of
tail-anchored membrane proteins into the endoplasmic reticulum membrane
in mammalian cells. Mol Cell 48:387–397.
doi:10.1016/j.molcel.2012.08.028
Yang J, Anishchenko I, Park H, Peng Z, Ovchinnikov S, Baker D. 2020.
Improved protein structure prediction using predicted interresidue
orientations. Proc Natl Acad Sci U S A
117:1496–1503. doi:10.1073/pnas.1914677117
Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. 2015. The I-TASSER
Suite: Protein structure and function prediction. Nat
Methods 12:7–8. doi:10.1038/nmeth.3213
Yang ZR, Thomson R, McNeil P, Esnouf RM. 2005. RONN: The
bio-basis function neural network technique applied to the detection of
natively disordered regions in proteins. Bioinformatics
21:3369–3376. doi:10.1093/bioinformatics/bti534
Yariv B, Yariv E, Kessel A, Masrati G, Chorin AB, Martz E, Mayrose I,
Pupko T, Ben-Tal N. 2023. Using evolutionary data to make sense of
macromolecules with a “face-lifted” ConSurf.
Protein Science 32:e4582. doi:10.1002/pro.4582
Yu G. 2022. Data integration, manipulation and visualization of
phylogenetic trees. CRC Press.
Yu G. 2020. Using ggtree to Visualize Data on
Tree-Like Structures. Curr Protoc Bioinformatics
69:e96. doi:10.1002/cpbi.96
Yuan S, Qin W, Riordan CR, McSwiggin H, Zheng H, Yan W. 2015. Ubqln3, a
testis-specific gene, is dispensable for embryonic development and
spermatogenesis in mice. Mol Reprod Dev
82:266–267. doi:10.1002/mrd.22475
Zadeh JN, Steenberg CD, Bois JS, Wolfe BR, Pierce MB, Khan AR, Dirks RM,
Pierce NA. 2011. NUPACK: Analysis and design
of nucleic acid systems. Journal of Computational Chemistry
32:170–173. doi:10.1002/jcc.21596
Zanata SM, Lopes MH, Mercadante AF, Hajj GNM, Chiarini LB, Nomizo R,
Freitas ARO, Cabral ALB, Lee KS, Juliano MA, de Oliveira E, Jachieri SG,
Burlingame A, Huang L, Linden R, Brentani RR, Martins VR. 2002.
Stress-inducible protein 1 is a cell surface ligand for cellular prion
that triggers neuroprotection. EMBO J
21:3307–3316. doi:10.1093/emboj/cdf325
Zeytuni N, Zarivach R. 2012. Structural and functional discussion of the
tetra-trico-peptide repeat, a protein interaction module.
Structure 20:397–405. doi:10.1016/j.str.2012.01.006
Zhang D, Raasi S, Fushman D. 2008. Affinity makes the difference:
Nonselective interaction of the UBA domain of
Ubiquilin-1 with monomeric ubiquitin and polyubiquitin
chains. J Mol Biol 377:162–180. doi:10.1016/j.jmb.2007.12.029
Zhao G, London E. 2006. An amino acid "transmembrane tendency" scale
that approaches the theoretical limit to accuracy for prediction of
transmembrane helices: Relationship to biological hydrophobicity.
Protein Sci 15:1987–2001. doi:10.1110/ps.062286306
Zhaxybayeva O, Gogarten JP, Charlebois RL, Doolittle WF, Papke RT. 2006.
Phylogenetic analyses of cyanobacterial genomes: Quantification of
horizontal gene transfer events. Genome Res
16:1099–1108. doi:10.1101/gr.5322306
Zhou J, Rudd KE. 2013. EcoGene 3.0. Nucleic Acids
Research 41:D613–624. doi:10.1093/nar/gks1235
Zhou T, Radaev S, Rosen BP, Gatti DL. 2000. Structure of the ArsA
ATPase: The catalytic subunit of a heavy metal resistance pump.
EMBO J 19:4838–4845. doi:10.1093/emboj/19.17.4838
Zientara-Rytter K, Subramani S. 2019. The Roles of
Ubiquitin-Binding Protein Shuttles in the Degradative
Fate of Ubiquitinated Proteins in the
Ubiquitin-Proteasome System and Autophagy.
Cells 8:40. doi:10.3390/cells8010040
Zimorski V, Ku C, Martin WF, Gould SB. 2014. Endosymbiotic theory for
organelle origins. Curr Opin Microbiol
22:38–48. doi:10.1016/j.mib.2014.09.008
Zopf D, Bernstein HD, Johnson AE, Walter P. 1990. The methionine-rich
domain of the 54 kd protein subunit of the signal recognition particle
contains an RNA binding site and can be crosslinked to a
signal sequence. EMBO J 9:4511–4517. doi:10.1002/j.1460-2075.1990.tb07902.x