2 Molecular basis of tail-anchored integral membrane protein recognition by the co-chaperone Sgt2
Adapted from Lin K-F, Fry MY*, Saladi SM*, Clemons WM. 2021. Molecular basis of tail-anchored integral membrane protein recognition by the co-chaperone Sgt2. J Biol Chem 296:100441. doi:10.1016/j.jbc.2021.100441
2.1 Abstract
The targeting and insertion of tail-anchored (TA) integral membrane proteins (IMP) into the correct membrane is critical for cellular homeostasis. The fungal protein Sgt2, and its human homolog SGTA, is the entry point for clients to the Guided Entry of Tail-anchored protein (GET) pathway, which targets ER-bound TA IMPs. Consisting of three structurally independent domains, the C-terminus of Sgt2 binds to the hydrophobic transmembrane domain (TMD) of clients. However, the exact binding interface within Sgt2 and molecular details that underlie its binding mechanism and client preference are not known. Here, we reveal the mechanism of Sgt2 binding to hydrophobic clients, including TA IMPs. Through sequence analysis, biophysical characterization, and a series of capture assays, we establish that the Sgt2 C-terminal domain is flexible but conserved, and sufficient for client binding. A molecular model for this domain reveals a helical hand forming a hydrophobic groove approximately 15Å long, that is consistent with our observed higher affinity for client TMDs with a hydrophobic face and a minimal length of 11 residues. This work places Sgt2 into a broader family of TPR-containing co-chaperone proteins, demonstrating structural and sequence-based similarities to the DP domains in the yeast Hsp90 and Hsp70 coordinating protein, Sti1.
Keywords: Tail-anchored proteins, Protein targeting, co-chaperones, Sti1, Hop
2.2 Introduction
An inherently complicated problem of cellular homeostasis is the biogenesis of hydrophobic IMPs which are synthesized in the cytoplasm and must be targeted and inserted into a lipid bilayer. Accounting for ~25% of transcribed genes (Pieper et al., 2013), IMPs are primarily targeted by cellular signal binding factors that recognize a diverse set of hydrophobic \(\alpha\)-helical signals as they emerge from the ribosome (Aviram and Schuldiner, 2017; Guna and Hegde, 2018; Shao and Hegde, 2011a). One important class of IMPs are tail-anchored (TA) proteins whose hydrophobic signals are their single helical transmembrane domain (TMD) located near the C-terminus and are primarily targeted post-translationally to either the ER or mitochondria (Chartron et al., 2012a; Denic, 2012; Hegde and Keenan, 2011; Kutay et al., 1993; Wattenberg and Lithgow, 2001). In the case of the canonical pathway for ER-destined TA IMPs, each is first recognized by homologs of mammalian SGTA (small glutamine tetratricopeptide repeat protein) (Chio et al., 2017; Guna and Hegde, 2018; Hegde and Keenan, 2011; Shao and Hegde, 2011b). Common to all signal binding factors is the need to recognize, bind, and then hand off a hydrophobic helix. How such factors can maintain specificity to a diverse set of hydrophobic clients that must subsequently be released remains an important question.
Homologs of Saccharomyces cerevisiae Sgt2 (ySgt2) and Homo sapiens SGTA (referred to here as hSgt2 and collectively Sgt2 for simplicity), are involved in a variety of cellular processes regarding the homeostasis of membrane proteins including the targeting of TA IMPs (Chartron et al., 2012a, 2011; Simon et al., 2013; Wang et al., 2010), retrograde transport of membrane proteins for ubiquitination and subsequent proteasomal degradation(Xu et al., 2012), and regulation of mislocalized membrane proteins (MLPs) (Leznicki and High, 2012; Wunderley et al., 2014). Among these, the role of Sgt2 in the primary pathways responsible for targeting TA clients to the endoplasmic reticulum (ER) is best characterized, i.e., the fungal Guided Entry of Tail-anchored proteins (GET) or the mammalian Transmembrane Recognition Complex (TRC) pathway. In the GET pathway, Sgt2 functions by binding a cytosolic TA client then transferring the TA client to the ATPase chaperone Get3 (human homolog is also Get3) with the aid of the heteromeric Get4/Get5 complex (human Get4/Get5/Bag6 complex) (Gristick et al., 2014; Mock et al., 2015; Wang et al., 2011, 2010). In this process, TA client binding to Sgt2, after hand-off from Hsp70, is proposed as the first committed step to ensure that ER TA clients are delivered to the ER membrane while mitochondrial TA clients are excluded (Cho and Shan, 2018; Shao and Hegde, 2011a; Wang et al., 2010). Subsequent transfer of the TA client from Sgt2 to the ATP bound Get3 induces conformational changes in Get3 that trigger ATP hydrolysis, releasing Get3 from Get4 and favoring binding of the Get3-TA client complex to the Get1/2 receptor at the ER leading to release of the TA client into the membrane (Rome et al., 2014; Schuldiner et al., 2008; Stefer et al., 2011; Vilardi et al., 2011; Yamamoto and Sakisaka, 2012). Deletions of yeast GET genes (i.e., get1\(\Delta\), get2\(\Delta\), or get3\(\Delta\)) cause cytosolic aggregation of TA clients dependent on Sgt2 (Kiktev et al., 2012; Schuldiner et al., 2008).
In addition to targeting TA IMPs, there is evidence hSgt2 promotes degradation of IMPs through the proteasome by cooperating with the Bag6 complex, a heterotrimer containing Bag6, hGet4, and hGet5, which acts as a central hub for a diverse physiological network related to protein targeting and quality control (Hessa et al., 2011; Mock et al., 2015; Rodrigo-Brenni et al., 2014; Xu et al., 2013). The Bag6 complex can associate with ER membrane-embedded ubiquitin regulatory protein UbxD8, transmembrane protein gp78, proteasomal component Rpn10c, and an E3 ubiquitin protein ligase RNF126 thereby connecting hSgt2 to ER associated degradation (ERAD) and proteasomal activity. Depletion of hSgt2 significantly inhibits turnover of ERAD IMP clients and elicits the unfolded protein response (Wunderley et al., 2014). Furthermore, the cellular level of MLPs in the cytoplasm could be maintained by co-expression with hSgt2, which possibly antagonize ubiquitination of MLPs to prevent proteasomal degradation (Leznicki and High, 2012; Xu et al., 2012). These studies demonstrate an active role of hSgt2 in triaging IMPs in the cytoplasm and the breadth of hSgt2 clients including TA IMPs, ERAD, and MLPs all harboring one or more TMD. Roles for hSgt2 in disease include polyomavirus infection(Dupzyk et al., 2017), neurodegenerative disease (Kiktev et al., 2012; Long et al., 2012), hormone-regulated carcinogenesis (Buchanan et al., 2007; Trotta et al., 2013), and myogenesis(Wang et al., 2003), although the underlying molecular mechanisms are still unclear.
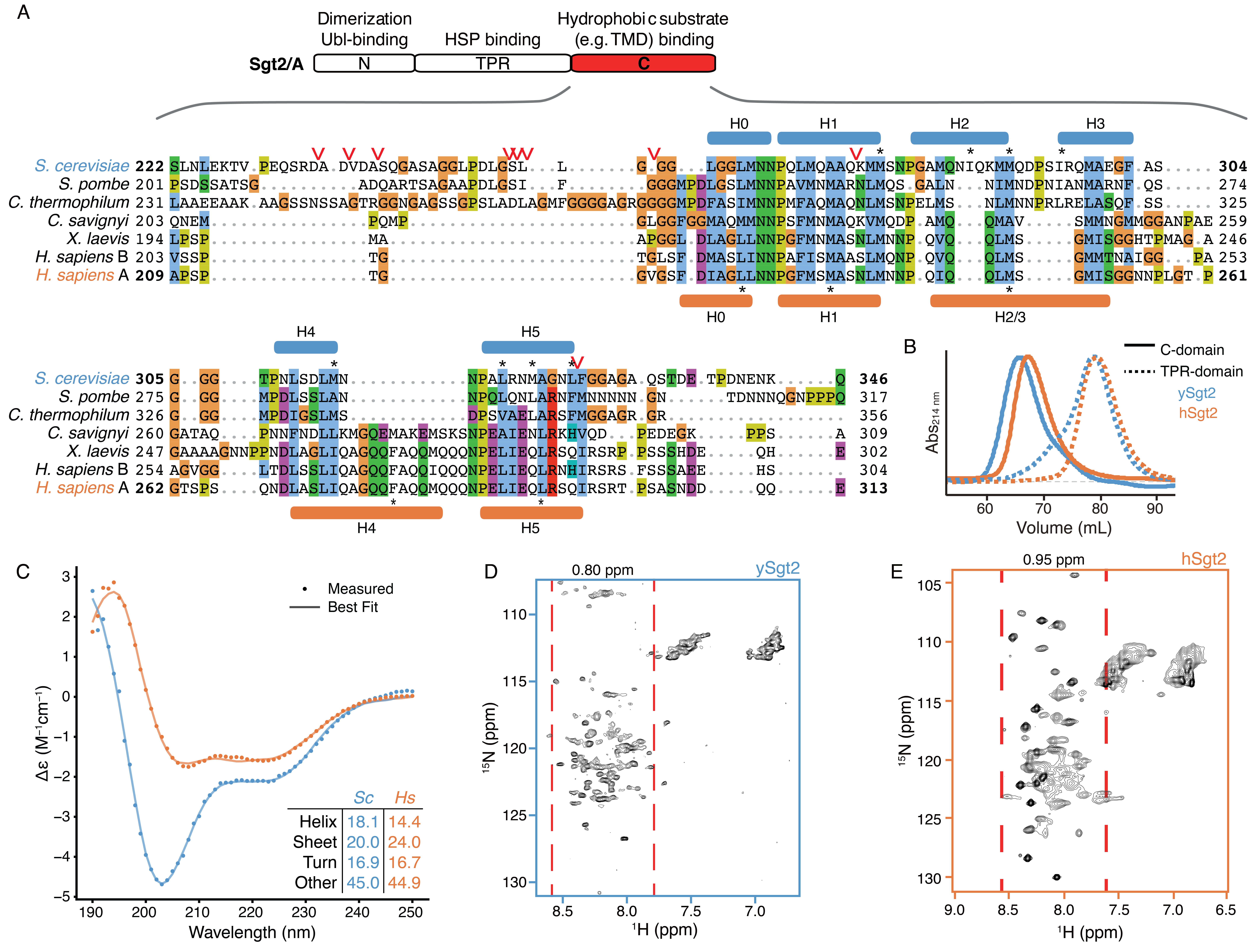
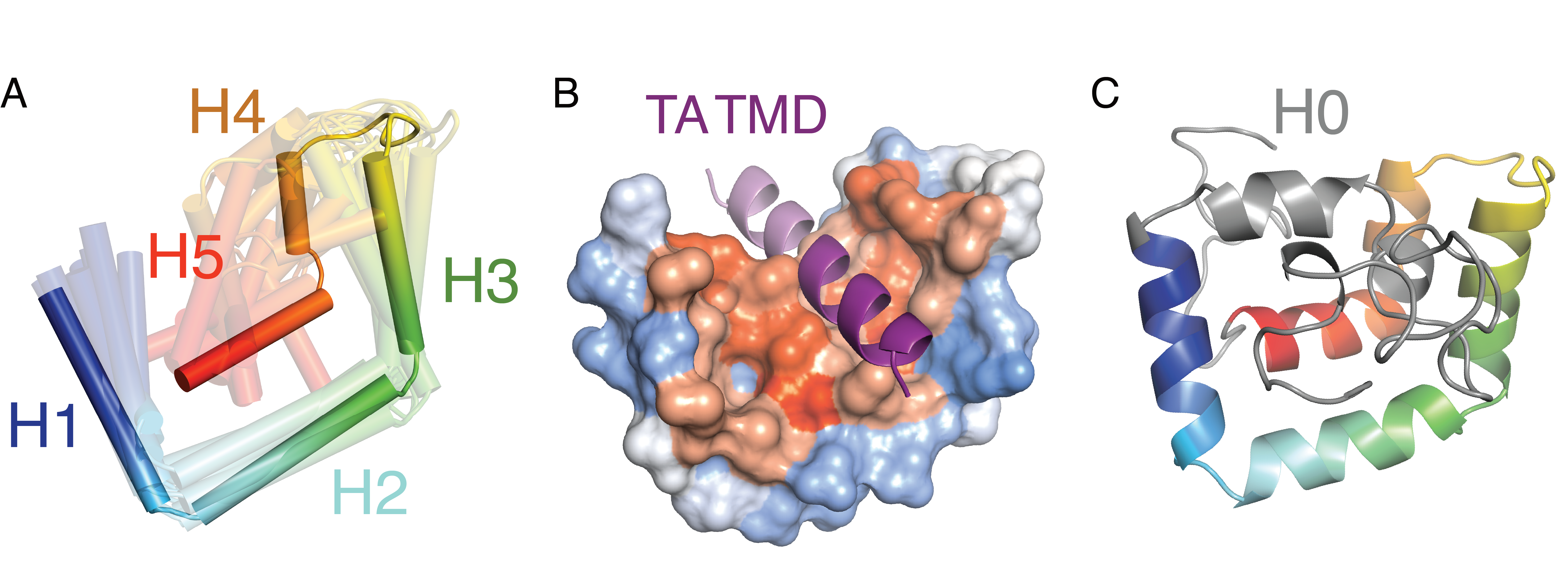
The architecture of Sgt2 includes three structurally independent domains that define the three different interactions of Sgt2 (Figure 2.1A) (Callahan et al., 1998; Chartron et al., 2012b, 2011; Cziepluch et al., 1998; Liou and Wang, 2005). The N-terminal domain forms a homo-dimer composed of a four-helix bundle with 2-fold symmetry that primarily binds to the ubiquitin-like domain (UBL) of Get5/Ubl4A for TA IMP targeting (Chartron et al., 2012b; Winnefeld et al., 2006) or interacts with the UBL on the N-terminal region of Bag6 (Darby et al., 2014) where it is thought to initiate downstream degradation processes (Rodrigo-Brenni et al., 2014; Xu et al., 2013, 2012). The central region comprises a co-chaperone domain with three repeated TPR motifs arranged in a right handed-superhelix forming a ‘carboxylate clamp’ for binding the C-terminus of heat-shock proteins (HSP) (Chartron et al., 2011; Dutta and Tan, 2008). The highly conserved TPR domain was demonstrated to be critical in modulating propagation of yeast prions by recruiting HSP70 (Kiktev et al., 2012) and may associate with the proteasomal factor Rpn13 to regulate MLPs (Leznicki et al., 2015). More recently, it was demonstrated that mutations to residues in the TPR domain which prevent Hsp70 binding impair the loading of TA clients onto ySgt2 (Cho and Shan, 2018), consistent with a direct role of Hsp70 in TA IMP targeting via the TPR domain. The C-terminal methionine-rich domain of Sgt2 is responsible for binding to hydrophobic clients such as TA IMPs (Liou and Wang, 2005; Shao and Hegde, 2011b; Waheed et al., 2016). Other hydrophobic segments have been demonstrated to interact with this domain such as the membrane protein Vpu (viral protein U) from human immunodeficiency virus type-1 (HIV-1), the TMD of tetherin (Waheed et al., 2016), the signal peptide of myostatin (Wang et al., 2003), and the N-domain of the yeast prion forming protein Sup35 (Kiktev et al., 2012). All of these studies suggest that the C-terminus of Sgt2 binds broadly to hydrophobic stretches, yet structural and mechanistic information for client recognition is lacking.
In this study, we provide the first structural characterization of the C-domains from Sgt2 (Sgt2-C) and show that, in the absence of substrate, it is relatively unstructured. We demonstrate that a conserved region of the C-domain, defined here as Ccons, is sufficient for client binding. Analysis of the Ccons sequence identifies six amphipathic helices whose hydrophobic residues are required for client binding. Based on this, we computationally generate an ab initio structural model that is validated by point mutants and disulfide crosslinking. Artificial clients are then used to define the properties within clients critical for binding to Sgt2-C. The results show that Sgt2-C falls into a larger STI1 family of TPR-containing co-chaperones and allow us to propose a mechanism for client binding.
2.3 Results
2.3.1 The flexible Sgt2-C domain
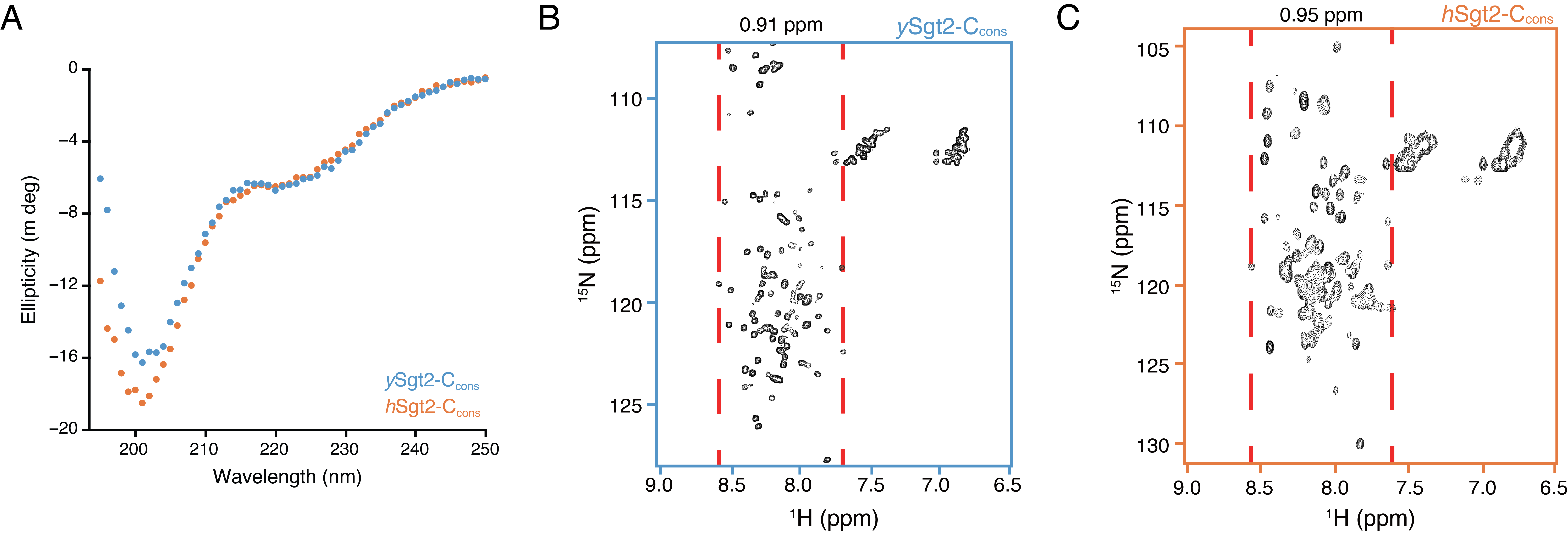
Based on sequence alignment (Figure 2.1A), the Sgt2-C contains a conserved core of six predicted helices flanked by unstructured loops that vary in length and sequence. Previous experimental work suggested that this region is particularly flexible, as this domain in the Aspergillus fumigatus homolog is sensitive to proteolysis (Chartron et al., 2011). Similarly, for ySgt2-TPR-C, the sites sensitive to limited proteolysis primarily occur within the loops flanking the conserved helices (Figure 2.1A, red arrows* and Supplementary Figure 2.1B). This flexible nature of the C-domain likely contributes to its anomalous passage through a gel-filtration column where Sgt2-C elutes much earlier than the similarly-sized, but well-folded, Sgt2 TPR-domain (Figure 2.1B), as is typical for unstructured proteins (Graether, 2018). The larger hydrodynamic radius matches previous small-angle X-ray scattering measurement of the ySgt2 TPR-C domain that indicated a partial unfolded characteristic in a Kratky plot analysis. The circular dichroism (CD) spectra for both homologs suggests that the C-domain and a predicted six \(\alpha\)-helical methionine-rich region of Sgt2-C (Figure 2.1A), hereafter referred to as Sgt2-Ccons, largely assume a random-coil conformation, with 40-45% not assignable to a defined secondary structure category (Figure 2.1C, Supplementary Figure 2.1A) (Luo and Baldwin, 1997). The well-resolved, sharp, but narrowly dispersed chemical shifts of the backbone amide protons in 1H-15N HSQC spectra of Sgt2-C (Figure 2.1D,E) and Sgt2-Ccons (Supplementary Figure 2.1B,C), indicate a significant degree of backbone mobility, similar to natively unfolded proteins (Dyson and Wright, 2004) and consistent with results seen by others (Martínez-Lumbreras et al., 2018), further highlighting the lack of stable tertiary structure (Chartron et al., 2011). Taken all together, Sgt2-C appears to be a flexible domain.
2.3.2 The conserved region of the C-domain is sufficient for substrate binding
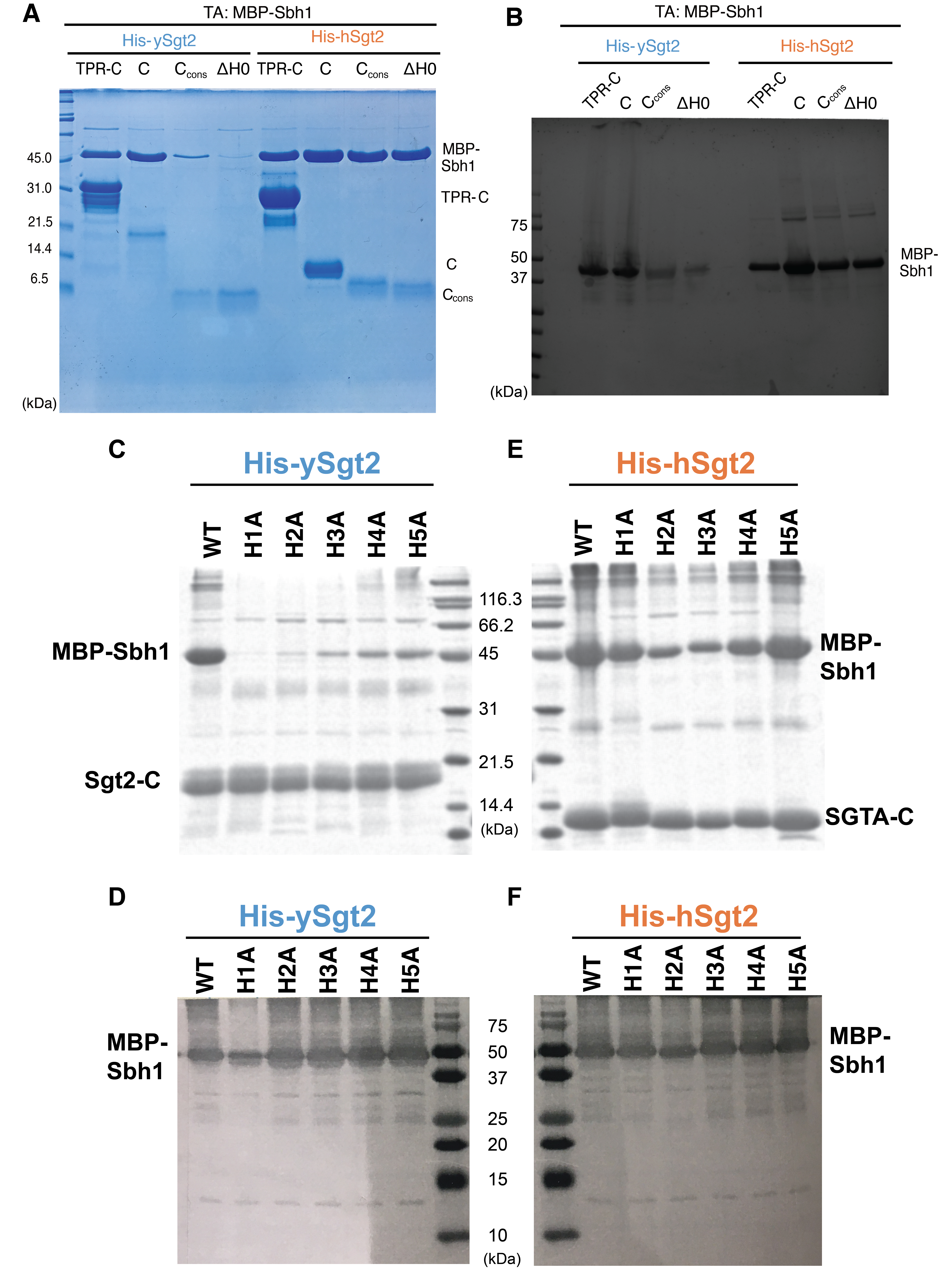
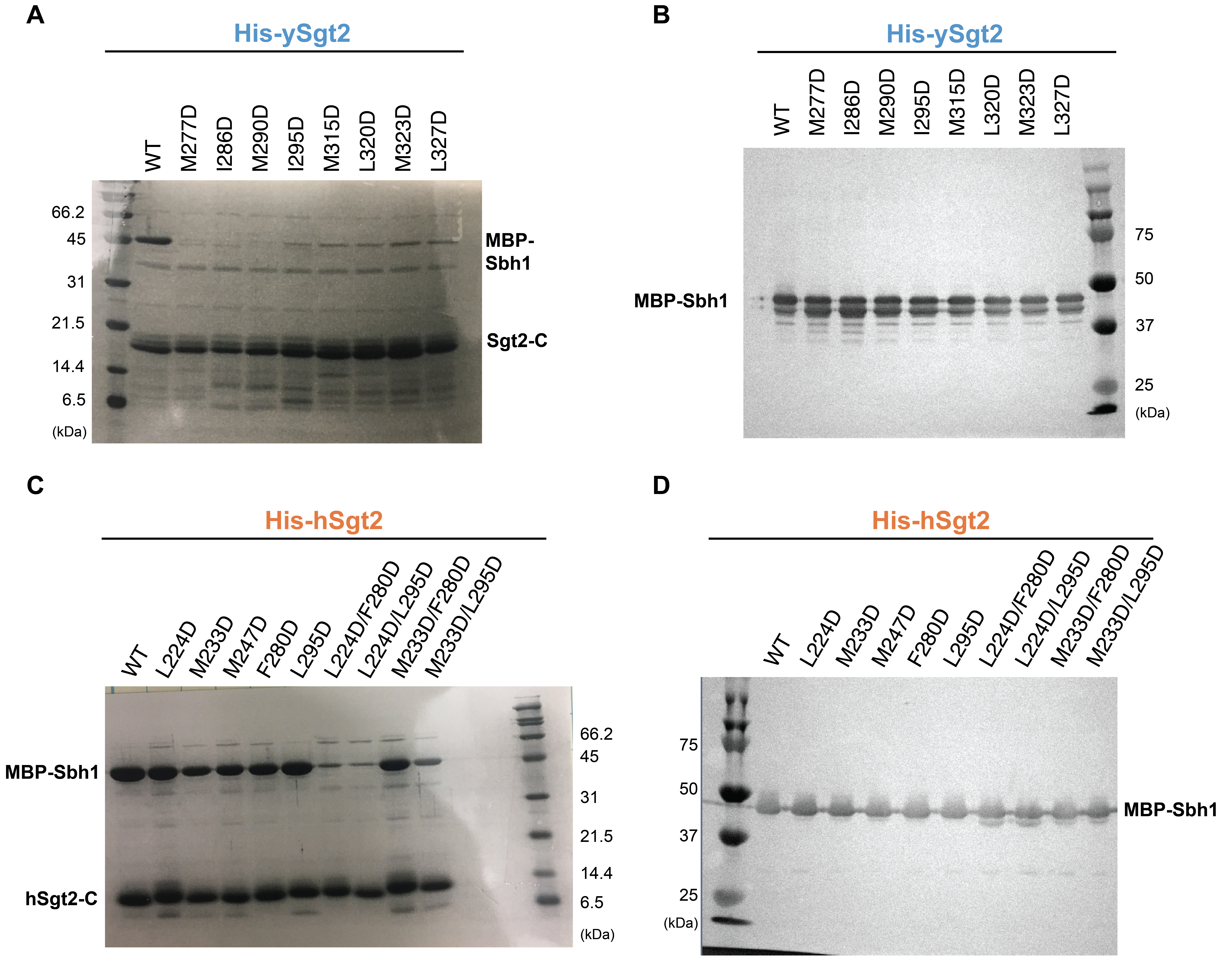
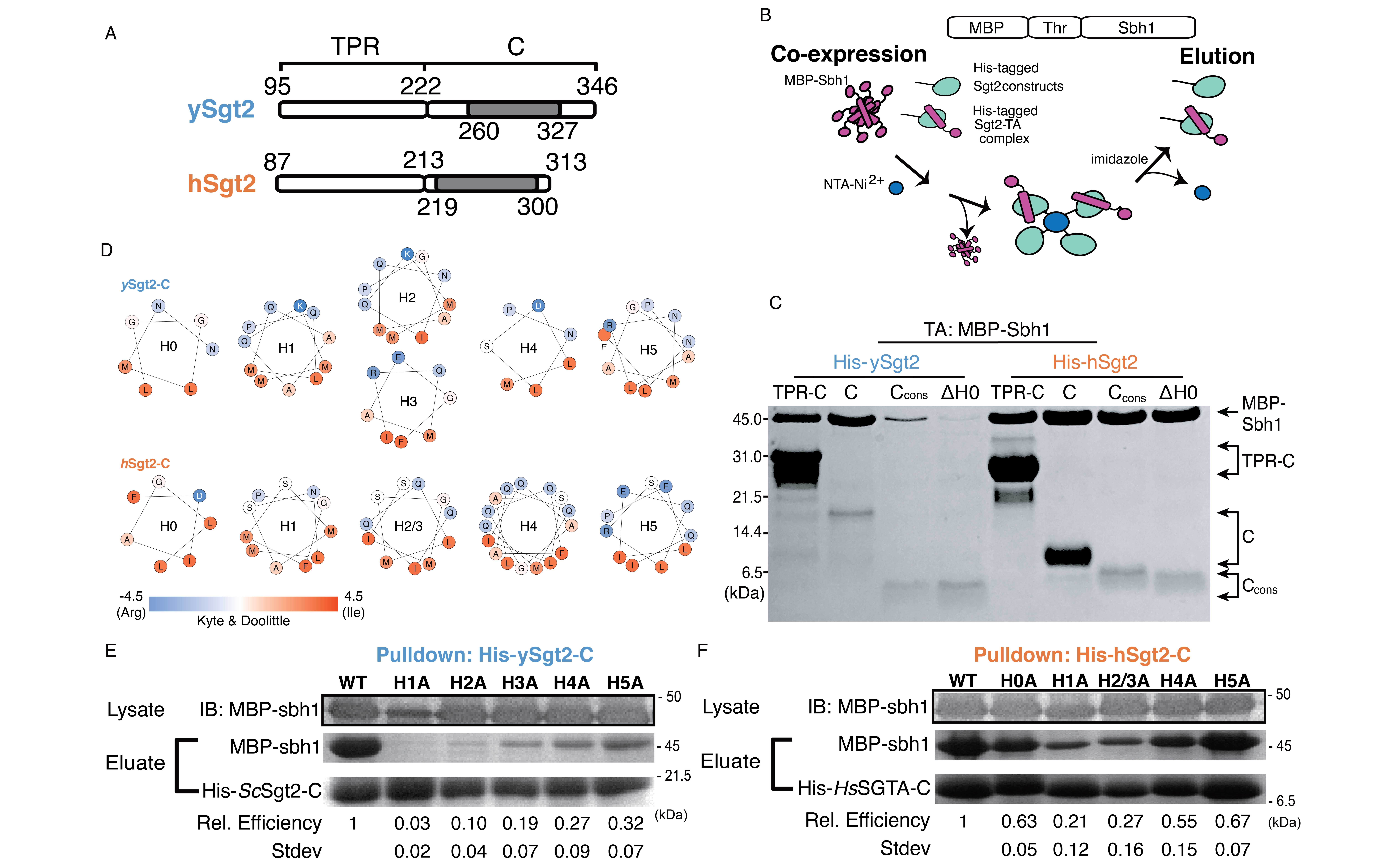
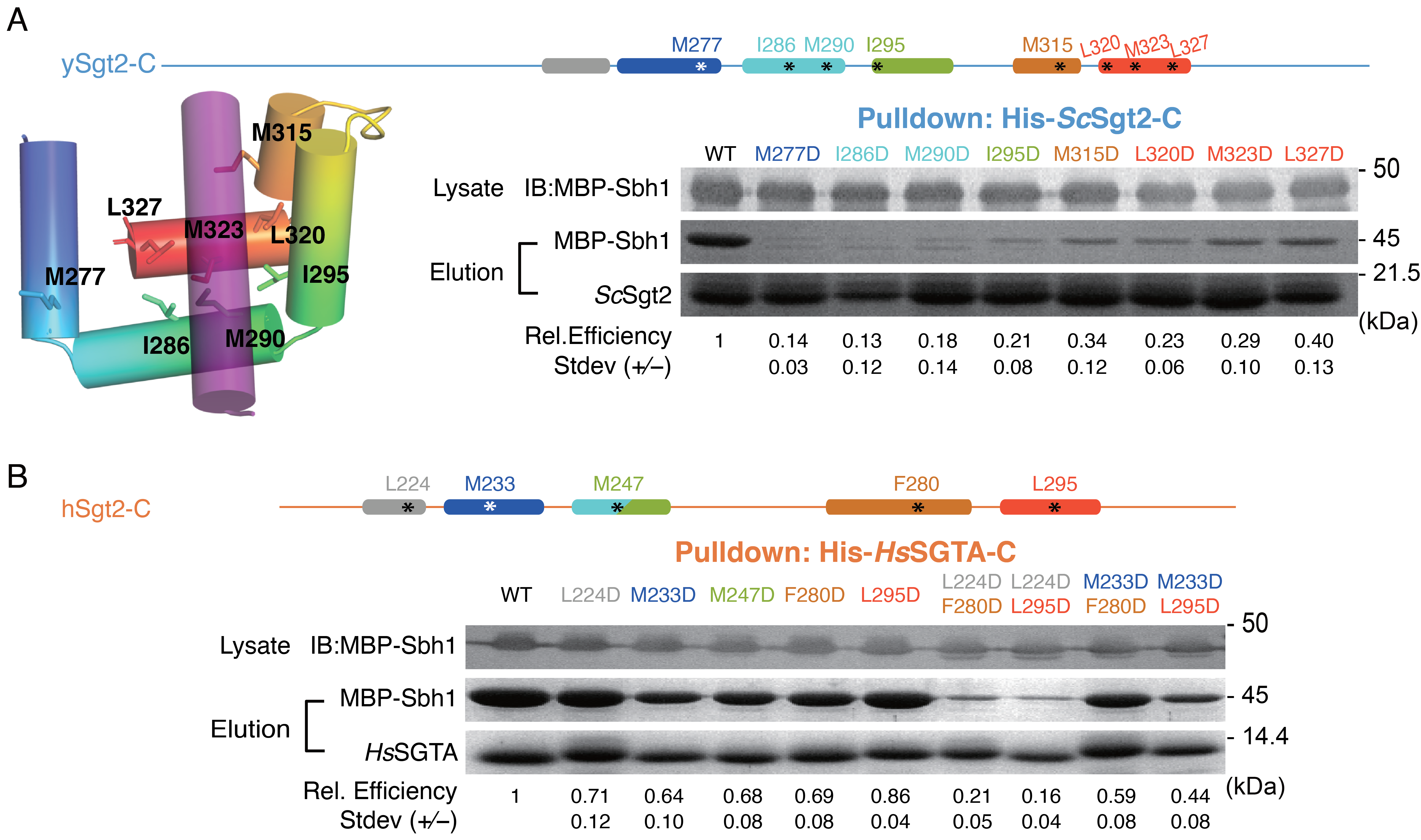
We then asked if the flexible Sgt2-C is the site of client binding in the co-chaperone and if so, where within this domain is the binding region. During purification Sgt2-C is susceptible to proteolytic activity being cut at several specific sites (Figure 2.1A). Proteolysis occurred primarily at Leu327 and in the poorly conserved N-terminal region (between Asp235-Gly258). Given the intervening region (ySgt2 G258-L327) is conserved (Figure 2.1A), it and the corresponding region in hSgt2 may mediate client binding (Figure 2.2A, grey). To test this, we established a set of his-tagged Sgt2 constructs of various lengths (Figure 2.2C, Supplementary Figure 2.2A). These Sgt2-C truncations were co-expressed with an MBP-tagged client, Sbh1, and binding was detected by the presence of captured TA clients in nickel elution fractions (Figure 2.2B). The TA protein Sbh1 is the yeast homolog of the mammalian Sec61-gamma, a component of the ER-resident Sec translocon. While the relative efficiency of MBP-Sbh1 capture cannot be assessed in this assay due to differences in total protein levels (Supplementary Figure 2.2B), we can demonstrate the ability of a given construct to bind to the client. As previously seen(Wang et al., 2010), we confirm that Sgt2-TPR-C alone is sufficient for capturing a client (Figure 2.2C). As one might expect, the C-domain was also sufficient for binding the client. Interestingly, Sgt2-Ccons is sufficient for binding to Sbh1. Even a minimal region of the last 5 helices (referred to as \(\Delta\)H0) also captures Sbh1 (Figure 2.2C). The predicted helices in Sgt2-Ccons are amphipathic and their hydrophobic faces may be used for client binding (Figure 2.2D).
Each of the six helices in Sgt2-Ccons was mutated to replace the larger hydrophobic residues with alanines, dramatically reducing the overall hydrophobicity. For all of the helices, alanine replacement of the hydrophobic residues significantly reduces binding of Sbh1 to Sgt2-C (Figure 2.2E,F). While these mutants expressed at similar levels to the wild-type sequence, one cannot rule out that some of these changes may affect the tertiary structure of this domain. In general, these results imply that these amphipathic helices are necessary for client binding since removal of the hydrophobic faces disrupts binding. The overall effect on binding by each helix is different, with mutations in helices 1-3 having the most dramatic reduction in binding suggesting that these are more crucial for Sgt2-client complex formation. It is also worth noting, as this is a general trend, that hSgt2 is more resistant to mutations that affect binding (Figure 2.2F) than ySgt2, which likely reflect different thresholds for binding.
2.3.3 Molecular modeling of Sgt2-C domain
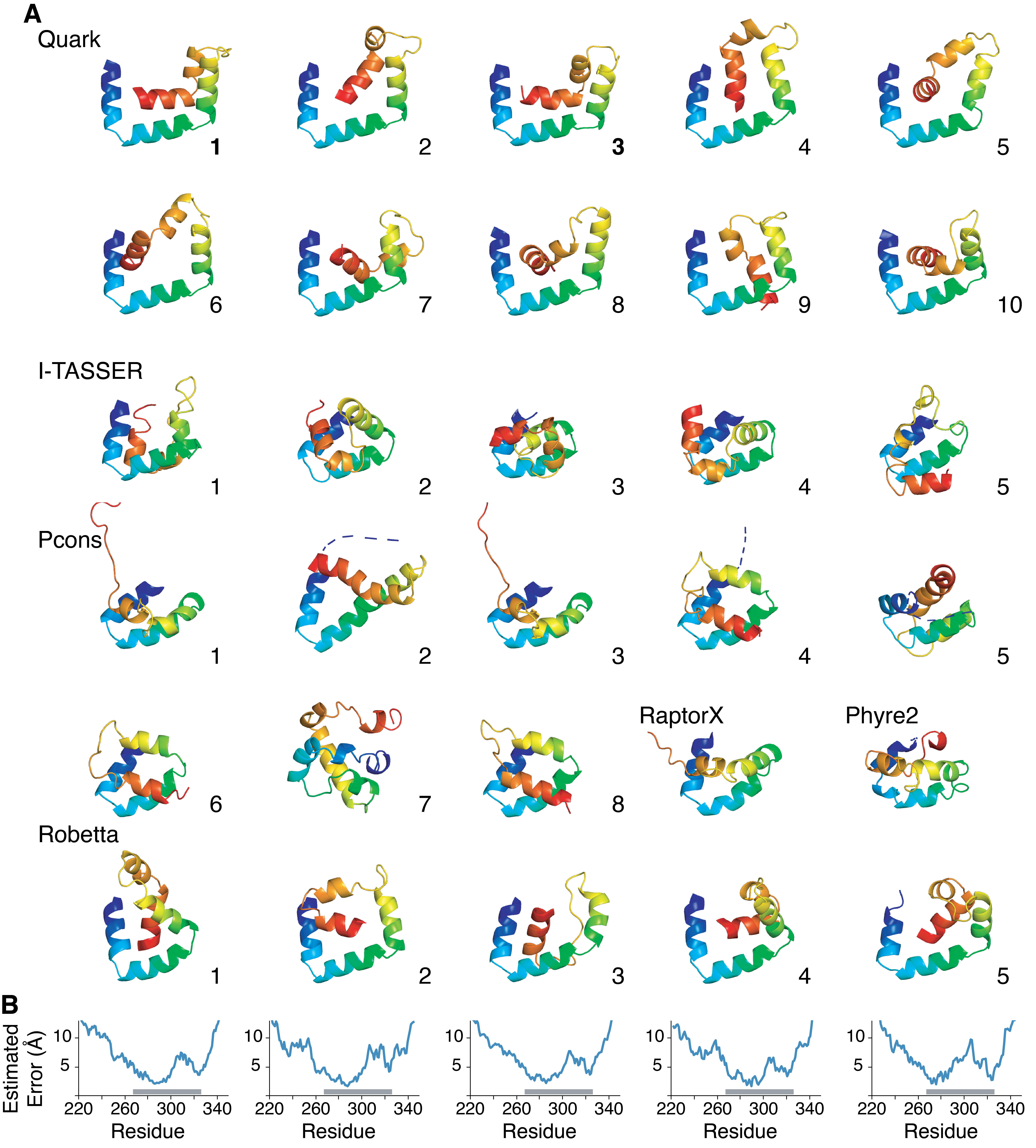
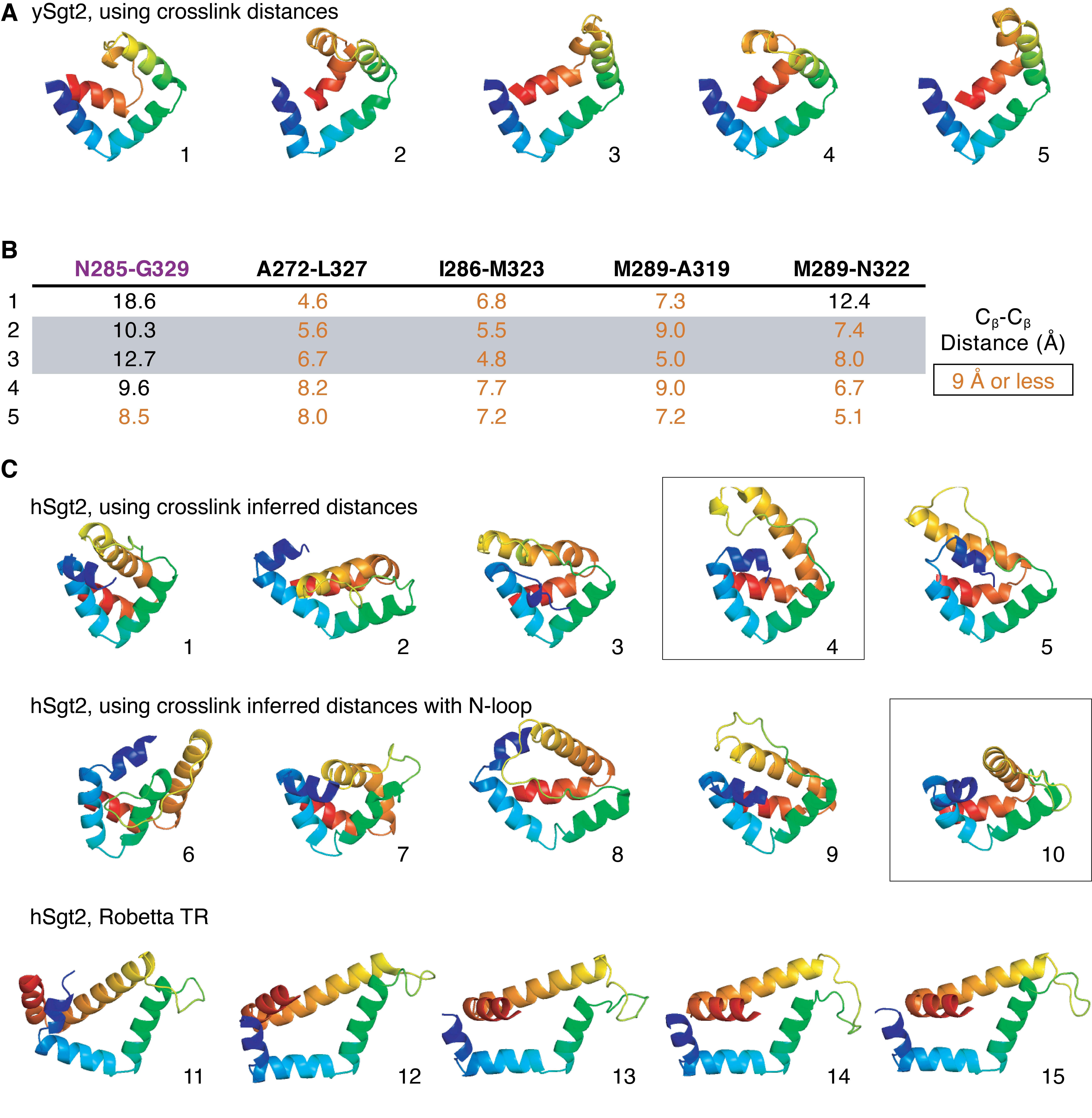
Despite the need for a molecular model, the C-domain has resisted structural studies, likely due to the demonstrated inherent flexibility. Based on the six conserved \(\alpha\)-helical amphipathic segments (Figure 2.1A) that contain hydrophobic residues critical for client binding (Figure 2.2D,E), we expect some folded structure to exist. Therefore, we performed ab initio molecular modeling of Sgt2-C using a variety of prediction methods resulting in a diversity of putative structures (Chio et al., 2019; Chun et al., 2012; Letunic et al., 2015; Shao et al., 2017; Xu and Zhang, 2012). As expected, all models showed buried hydrophobic residues as this is a major criterion for in silico protein folding. Residues outside the ySgt2-Ccons region adopted varied conformations consistent with their expected higher flexibility. Pruning these N- and C-terminal regions to focus on the ySgt2-Ccons region (Supplementary Figure 2.3A) revealed a potential binding interface for a hydrophobic substrate. Examples are seen in Quark models (1, 4, & 6 shown), Robetta 1 & 2, and I-TASSER 2 & 3, whereas others models had no clearly distinguishable groove. Given the intrinsic flexibility of the Sgt2-C domain, it is possible that models without a groove are found in the non-TMD bound structural ensemble.
For a working model of TMD-bound ySgt2-C, we chose the highest scoring Quark structures where a general consistent architecture is seen (Figure 2.3A) (Xu and Zhang, 2012). The overall model contained a potential client binding site, a hydrophobic groove formed by the amphipathic helices. The groove is approximately 15 Å long, 12 Å wide, and 10 Å deep, which is sufficient to accommodate three helical turns of an \(\alpha\)-helix, ~11 amino acids (Figure 2.3B).
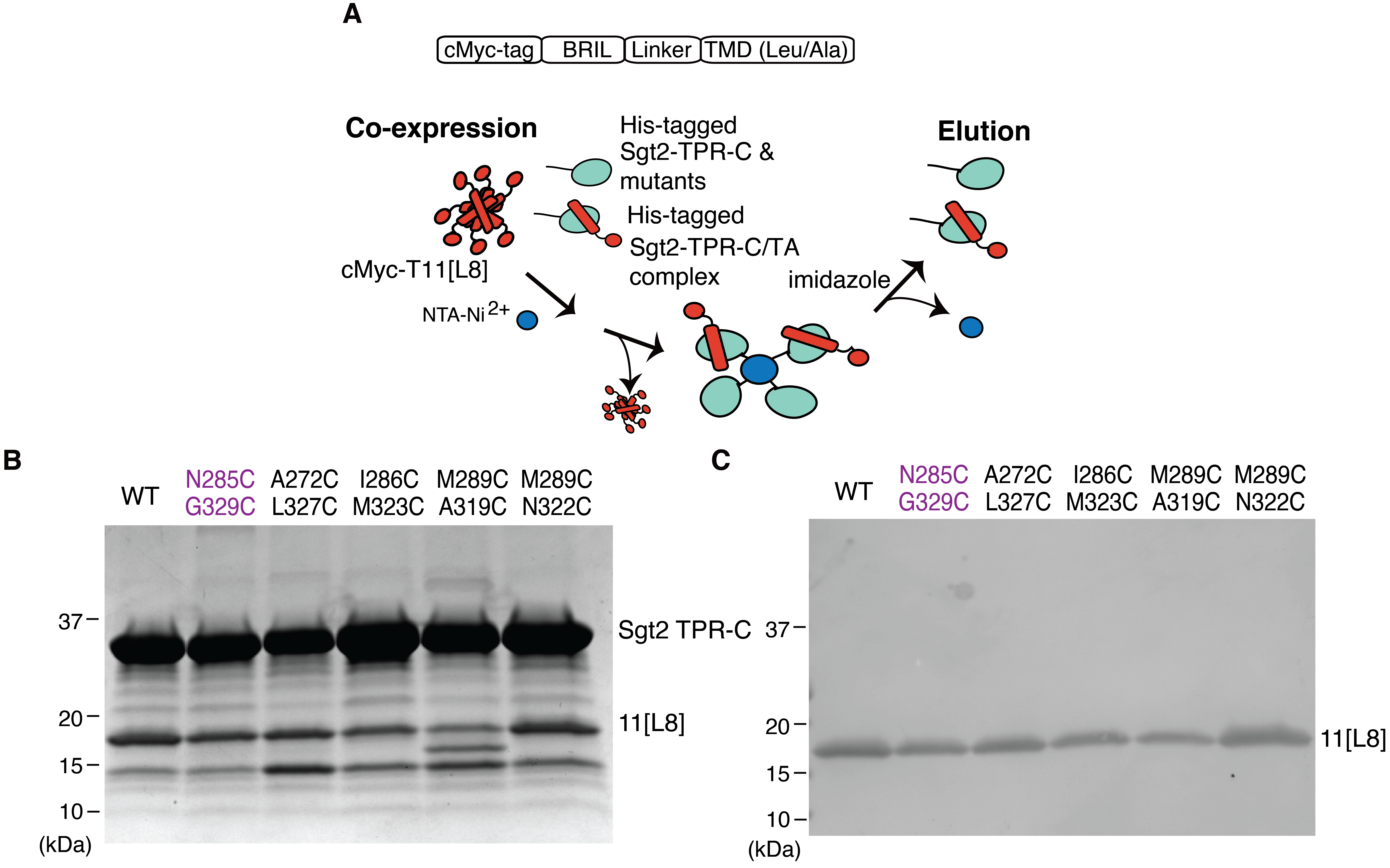
To validate the model, we interrogated the accuracy of the predicted structural arrangement by determining distance constraints from crosslinking experiments. We selected four pairs of residues in close spatial proximity and one pair far apart based on the Quark models (Figure 2.4A). Calculating a C\(\beta\)-C\(\beta\) distance between residue pairs for each model (Figure 2.4F), the Quark models 1 and 3 were the most consistent with an expected distance of 9Å or less for the close pairs. In all alternative models, the overall distances are much larger and should not be expected to form disulfide bonds in vitro if they represent a TMD-bound state. For Robetta, a number of the models have pairs of residues within 9Å and Robetta’s per-residue error estimate suggests relatively high confidence in the Ccons region (Supplementary Figure 2.3B).
As a control, we first confirmed that the cysteine-mutant pairs do not affect the function of ySgt2. We utilized an in vitro capture assay where a yeast Hsp70 homolog Ssa1 loaded with a TA client, Bos1, delivers the client to ySgt2 (Chio et al., 2019; Cho and Shan, 2018; Shao et al., 2017) (Figure 2.4C). Purified Ssa1 is mixed with detergent solubilized strep-tagged Bos1-TMD (a model ER TA client) that contained a p-benzoyl-l-phenylalanine (BPA) labeled residue, Bos1BPA, and diluted to below the critical micelle concentration resulting in soluble complexes of Bos1BPA/Ssa1. Full-length ySgt2 variants were each tested for the ability to capture Bos1BPA from Ssa1. After the transfer reaction, each was UV-treated to generate Bos1 crosslinks. Successful capture of the TA clients by ySgt2 was detected for all cysteine variants using an anti-strep Western blot and the appearance of a Bos1BPA/ySgt2 crosslink band, suggesting the mutations do not affect the structure or function of ySgt2 (Figure 2.4C).
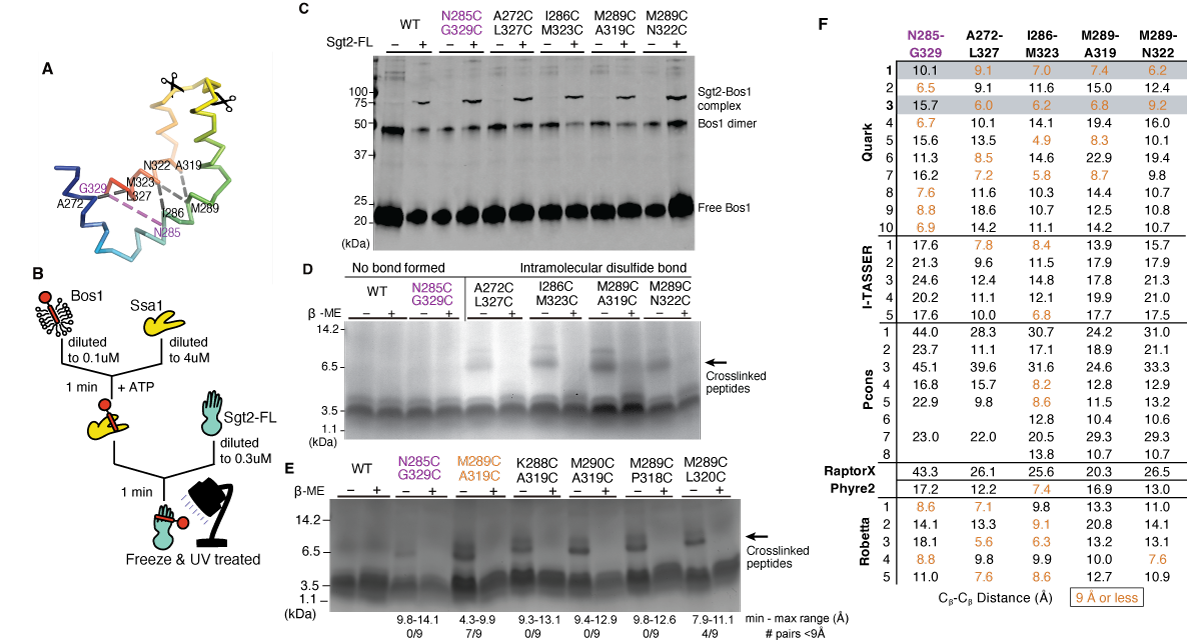
We and others have demonstrated that a monomeric Sgt2 is sufficient for binding to clients (Wang et al., 2010). For the distance experiment, each of the cysteine-mutant pairs was made in the more stable monomeric variant ySgt2-TPR-C. Each variant was coexpressed with an artificial client - a cMyc-tagged BRIL (small, 4-helix bundle protein used in previous work to aid in the crystallization of GPCRs (Chun et al., 2012)) with a C-terminal TMD consisting of eight leucines and three alanines, denoted 11[L8], and purified via nickel-affinity chromatography in reducing buffer (Supplementary Figure 2.4A). All of the ySgt2 mutants bound to the client and behaved similar to the wild-type (cysteine-free) further suggesting the mutants did not perturb the native structure (Supplementary Figure 2.4B). For disulfide crosslink formation, each eluate was oxidized, digested using the protease Glu-C, and crosslinks were identified by the visualization of a reducing-agent sensitive ~7.7kDa fragment in gel electrophoresis (Figure 2.4D). For both the wild-type construct and in N285C/G329C, where the pairs are predicted from the Quark models to be too distant for disulfide bond formation, no higher molecular weight band was observed. For the remaining pairs that are predicted to be close enough for bond formation, the 7.7kDa fragment was observed in each case and is labile in reducing conditions. Again, these results support the Ccons model derived from Quark.
With the four crosslinked pairs as distance constraints, new models were generated using Robetta with a restraint on the corresponding pairs of C\(\beta\) atoms less than 9Å (Supplementary Figure 2.5A). The Robetta models from these runs are similar to the top scoring models from Quark (Figure 2.3). Satisfyingly, the pair of residues that do not form disulfide crosslinks are generally consistent (Supplementary Figure 2.5B).
The improvement of the ySgt2 models predicted by Robetta with restraints included encouraged us to generate models for hSgt2-C with constraints. For this, pairs were defined based on sequence alignments of Sgt2 (Figure 2.1A) and used as restraints. The resulting predictions had architectures consistent with the equivalent regions predicted for ySgt2-Ccons, for example Robetta 4 (Supplementary Figure 2.5C, top). Although in general the predicted hSgt2 model is similar to that for ySgt2, the region that corresponds to H2 occupies a position that precludes a clear hydrophobic groove. For ySgt2, the longer N-terminal loop occupies the groove preventing the exposure of hydrophobics to solvent (Figure 2.3C, grey). For hSgt2, the shorter N-terminal loop may not be sufficient to similarly occupy the groove and allowing for the clear hydrophobic hand seen for the ySgt2-C. To correct for this, we replaced the sequence of the N-terminal loop of hSgt2-C with the ySgt2-C loop and ran structure prediction with the pairwise distance restraints. This resulted in a model where the loop occupies the groove and, when pruned away suggests the hydrophobic hand seen in yeast (Supplementary Figure 2.5C, middle boxed). Of note, we also generated models of hSgt2-C using the most recent Robetta method (transform-restrained) which produces new structures with a groove and similar helical-hand architecture across the board (Supplementary Figure 2.5C, bottom).
We sought to further test the robustness of our model considering the intrinsic flexibility of Sgt2-C by probing for disulfide bond formation with neighboring residues of one of our crosslinking pairs. While the C\(\beta\)-C\(\beta\) distance puts these adjacent pairs at farther than 9Å, mutating residues to cystines and measuring S-S distances across all possible pairs of rotamers provides a wider interval on possible distances and, therefore, the likelihood a disulfide bond will form (Figure 2.4E). Cysteine mutants were introduced to the residues adjacent to M289 and A319 in ySgt2-TPR-C resulting in four additional pairs: K288C/A319C, M290C/A319C, M289C/P318C, and M289C/L320C. As described previously, these mutants were coexpressed with a substrate, in this case the cMyc-tag was replaced with an MBP-tag. The MBP-tag on the artificial client allows for tandem amylose- and nickel-affinity chromatography to ensure eluates contained only Sgt2-TPR-C bound to substrate. Disulfide bond formation was conducted as before and a reductant sensitive band at 7.7kDa is observed for each of these adjacent pairs. While the geometry of each of these C-C pairs might suggest against disulfide bond formation, given the intrinsic flexibility of Sgt2-C, it is not surprising that each of these pairs are able to form disulfide bonds. As before, disulfide bond formation was detected for the M289C/A319C pair. In this new construct, we now see a small amount of disulfide bond formation in the distant N285C/G329C pair, likely an effect of switching to the MBP tag.
2.3.4 Structural similarity of Sgt2-C domain to STI1 domains
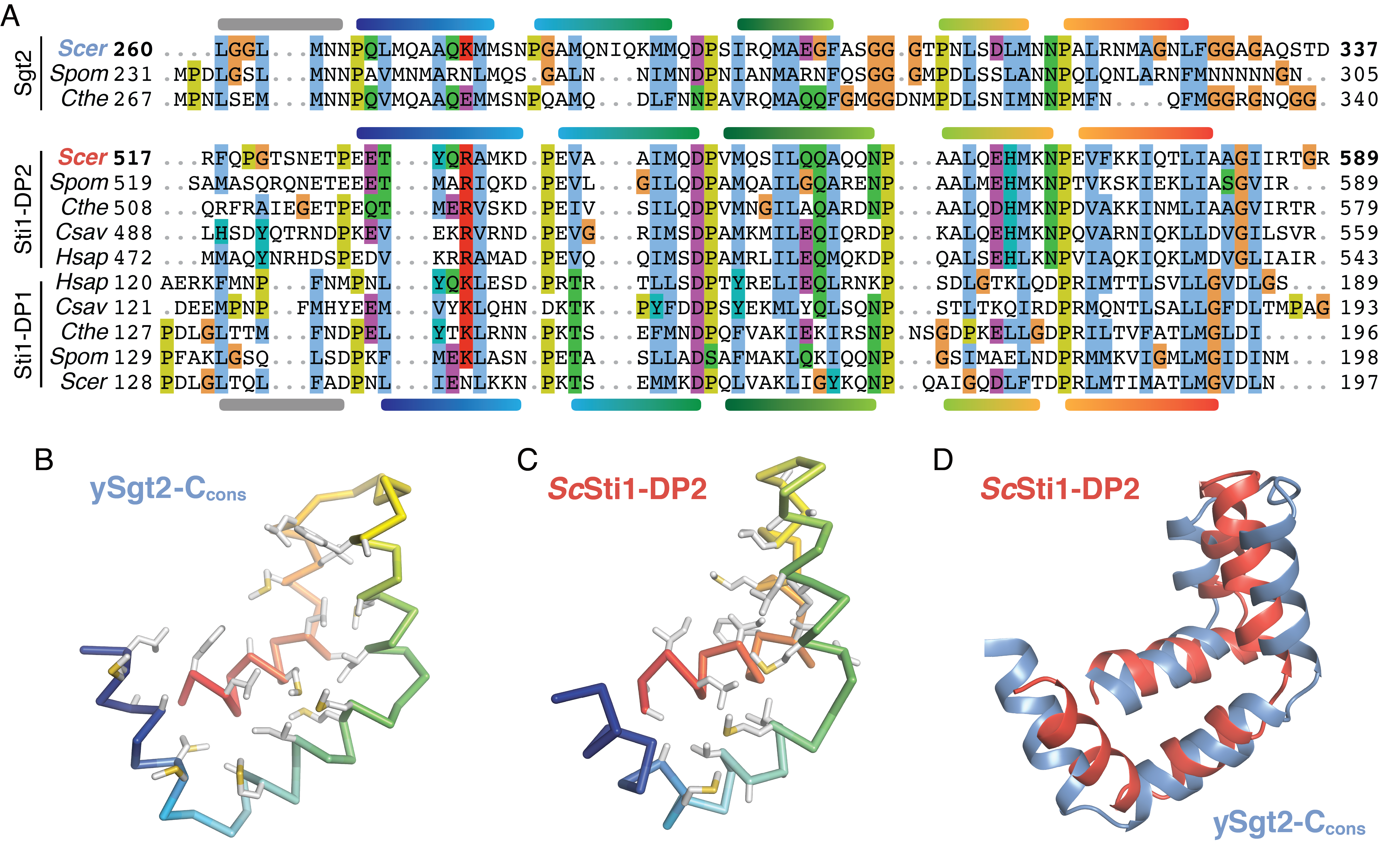
Attempts to glean functional insight for Sgt2-C from BLAST searches did not reliably return other families or non-Sgt2 homologs making functional comparisons difficult. A more extensive profile-based search using hidden Markov models from the SMART database (Letunic et al., 2015) identified a similarity to domains in the yeast co-chaperone Sti1 (HOP in mammals). First called DP1 and DP2, due to their prevalence of aspartates (D) and prolines (P), these domains have been shown to be required for client-binding by Sti1 (Li et al., 2013; Schmid et al., 2012) and are termed ‘STI1’-domains in bioinformatics databases (Letunic et al., 2015). In yeast Sti1 and its human homolog HOP (combined will be referred to here as Sti1), each of the two STI1 domains (DP1 and DP2) are preceded by Hsp70/90-binding TPR domains, similar to the domain architecture of Sgt2. Deletion of the second, C-terminal STI1-domain (DP2) from Sti1 in vivo is detrimental, impairing native activity of the glucocorticoid receptor (Schmid et al., 2012). In vitro, removal of the DP2 domain from Sti1 results in the loss of recruitment of the progesterone receptor to Hsp90 without interfering in Sti1-Hsp90 binding (Nelson et al., 2003). These results implicate DP2 in binding of Sti1 clients. In addition, others have noted that, broadly, STI1-domains may present a hydrophobic groove for binding the hydrophobic segments of a client(Li et al., 2013; Schmid et al., 2012). Furthermore, the similar domain organizations (i.e., Sgt2 TPR-C, Sti1 TPR-STI1) and molecular roles could imply an evolutionary relationship between these co-chaperones. Indeed, a multiple sequence alignment of the Sgt2-Ccons with several yeast STI1 domains (Figure 2.5A) reveals strong conservation of structural features. H1-H5 of the predicted helical regions in Ccons align directly with the structurally determined helices in the DP2 domain of Sti1; this includes complete conservation of helix breaking prolines and close alignment of hydrophobic residues in the amphipathic helices (Schmid et al., 2012).
Based on the domain architecture and homology, a direct comparison between the STI1 domain and Sgt2-Ccons can be made. A structure of DP2 solved by solution NMR reveals that the five amphipathic helices assemble to form a flexible helical-hand with a hydrophobic groove (Schmid et al., 2012). The lengths of the \(\alpha\)-helices in this structure concur with those inferred from the alignment in Figure 2.4A. Our molecular model of Sgt2-Ccons is strikingly similar to this DP2 structure (Figure 2.5B,C). An overlay of the DP2 structure and our molecular model demonstrates both Sgt2-Ccons and DP2 have similar lengths and arrangements of their amphipathic helices (Figure 2.5D). Consistent with our observations of flexibility in Sgt2-Ccons, Sti1-DP2 generates few long-range NOEs between its helices indicating that Sti1-DP2 also has a flexible architecture (Schmid et al., 2012). We consider this flexibility a feature of these helical-hands for reversible and specific binding of a variety of clients.
2.3.5 Binding mode of clients to Sgt2
We examined the Sgt2-Ccons surface that putatively interacts with clients by constructing hydrophobic-to-charge residue mutations that are expected to disrupt capture of clients by Sgt2. Similar to the helix mutations in Figure 2.2E,F, the capture assay was employed to establish the relative effects of individual mutations. A baseline was established based on the amount of the TA client Sbh1 captured by wild-type Sgt2-TPR-C. In each experiment, Sbh1 was expressed at the same level; therefore, differences in binding should directly reflect the affinity of Sgt2 mutants for clients. In all cases, groove mutations from hydrophobic to aspartate led to a reduction in client binding (Figure 2.6). The effects are most dramatic with ySgt2 where each mutant significantly reduced binding by 60% or more (Figure 2.6A). While all hSgt2 individual mutants saw a significant loss in binding, the results were more subtle with the strongest a ~36% reduction (M233D, Figure 2.6B). Double mutants were stronger with a significant decrease in binding relative to the individual mutants, more reflective of the individual mutants in ySgt2. As seen before (Figure 2.2E,F), we observe that mutations toward the N-terminus of Sgt2-C have a stronger effect on binding than those later in the sequence, whether single point mutants in the case of ySgt2 or double mutants for hSgt2.
2.3.6 Sgt2-C domain binds clients with a hydrophobic segment \(\geq\) 11 residues
With a molecular model for ySgt2-Ccons and multiple lines of evidence for a hydrophobic groove, we sought to better understand the specific requirements for TMD binding. TMD clients were designed where the overall (sum) and average (mean) hydrophobicity, length, and the distribution of hydrophobic character were varied in the TMDs. These artificial TMDs, a Leu/Ala helical stretch followed by a Trp, were constructed as C-terminal fusions to the soluble protein BRIL (Figure 2.7A). The total and mean hydrophobicity are controlled by varying the helix-length and the Leu/Ala ratio. For clarity, we define a syntax for the various artificial TMD clients to highlight the various properties under consideration: hydrophobicity, length, and distribution. The generic notation is TMD-length[number of leucines] which is represented, for example, as 18[L6] for a TMD of 18 amino acids containing six leucines.
Our first goal with the artificial clients was to define the minimal length of a TMD to bind to the C-domain. As described earlier in our single point mutation capture assays, captures of his-tagged Sgt2-TPR-C with the various TMD clients were performed. We define a relative binding efficiency as the ratio of captured TMD client by a Sgt2-TPR-C normalized to the ratio of a captured wild-type TA client by Sgt2-TPR-C. In this case we replaced the TMD in our artificial clients with the native TMD of Bos1 (Bos1TMD). The artificial client 18[L13] shows a comparable binding efficiency to Sgt2-TPR-C as that of Bos1TMD (Figure 2.7B). From the helical wheel diagram of the TMD for Bos1, we noted that the hydrophobic residues favored one face of the helix. We explored this ‘hydrophobic face’ by using model clients that maintained this orientation while shortening the length and maintaining the average hydrophobicity of 18[L13] (Figure 2.7B). Shorter helices of 14 or 11 residues, 14[L10] and 11[L8], also bound with similar affinity to Bos1. Helices shorter than 11 residues, 9[L6] and 7[L5], were not able to bind Sgt2-TPR-C (Figure 2.7B), establishing a minimal length of 11 residues for the helix, consistent with the dimensions of the groove predicted from the structural model (Figure 2.3).
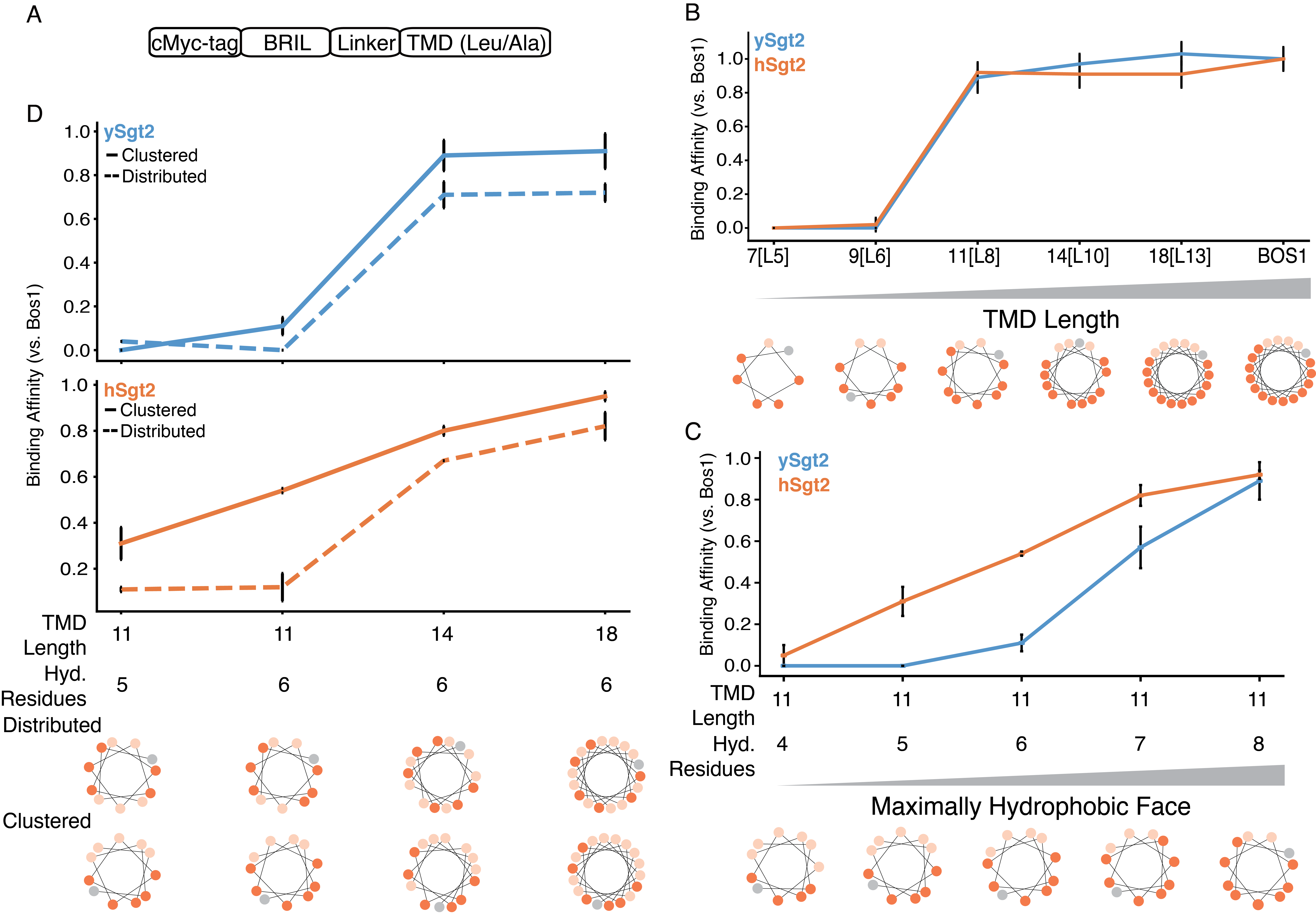
Since a detected binding event occurs with TMDs of at least 11 amino acids, we decided to probe this limitation further. The dependency of client hydrophobicity was tested by measuring complex formation of Sgt2-TPR-C and artificial TMD clients containing an 11 amino acid TMD with increasing number of leucines (11[Lx]). As shown in Figure 2.7C, increasing the number of leucines monotonically enhances complex formation, echoing previous results (Rao et al., 2016). hSgt2-TPR-C binds to a wider spectrum of hydrophobic clients than ySgt2-TPR-C, which could mean it has a more permissive hydrophobic binding groove, also reflected in the milder impact of Ala replacement and Asp mutations in hSgt2-TPR-C to TMD client binding (Figure 2.2F and Figure 2.6B).
2.3.7 Sgt2-C preferentially binds to TMDs with a hydrophobic face
Next, we address the properties within the TMD of clients responsible for Sgt2 binding. In the case of ySgt2, it has been suggested that the co-chaperone binds to TMDs based on hydrophobicity and helical propensity (Rao et al., 2016). In our system, our artificial TMDs consist of only alanines and leucines which have high helical propensities (Pace and Scholtz, 1998), and despite keeping the helical propensity constant and in a range that favors Sgt2 binding, there is still variation in binding efficiency. For the most part, varying the hydrophobicity of an artificial TMD client acts as expected, the more hydrophobic TMDs bind more efficiently to Sgt2-TPR-C (Figure 2.7C). Our Ccons model suggests the hydrophobic groove of ySgt2-C protects a TMD with highly hydrophobic residues clustered to one side (see Figure 2.3B). To test this, various TMD pairs with the same hydrophobicity, but different distributions of hydrophobic residues demonstrates TMD clients with clustered leucines have a higher relative binding efficiency than those with a more uniform distribution (Figure 2.7D). Helical wheel diagrams demonstrate the distribution of hydrophobic residues along the helix (e.g., bottom Figure 2.7D). The clustered leucines in the TMDs create a hydrophobic face which potentially interacts with the hydrophobic groove formed by the Sgt2-Ccons region, corresponding to the model in Figure 2.3B.
2.4 Discussion
Sgt2, the most upstream component of the GET pathway, plays a critical role in the targeting of TA IMPs to their correct membranes along with other roles in maintaining cellular homeostasis. Its importance as the first confirmed selection step of ER versus mitochondrial (Rao et al., 2016) destined TA IMPs necessitates a molecular model for client binding. Previous work demonstrated a role for the C-domain of Sgt2 to bind to hydrophobic clients, yet the exact binding domain remained to be determined. Through the combined use of biochemistry, bioinformatics, and computational modeling, we conclusively identify the minimal client-binding domain of Sgt2 and preferences in client binding. Here we present a validated structural model of the Sgt2 C-domain as a methionine-rich helical hand for grasping a hydrophobic helix and to provide a mechanistic explanation for binding a TMD of at least 11 hydrophobic residues.
Identifying the C-domain of Sgt2 as containing a STI1 places Sgt2 into a larger context of conserved co-chaperones (Figure 2.8A). In the co-chaperone family, the STI1 domains predominantly follow HSP-binding TPR domains connected by a flexible linker, reminiscent of the domain architecture of Sgt2. As noted above, for Sti1 these domains are critical for coordinated hand-off between Hsp70 and Hsp90 homologs (Röhl et al., 2015) as well as coordinating the simultaneous binding of two heat shock proteins. Both Sgt2 and the co-chaperone Hip coordinate pairs of TPR and STI1 domains by forming stable dimers via their N-terminal dimerization domains (Coto et al., 2018). With evidence for a direct role of the carboxylate-clamp in the TPR domain of Sgt2 for TA client-binding now clear (Cho and Shan, 2018), one can speculate that the two TPR domains may facilitate TA client entry into various pathways that use multiple heat shock proteins.
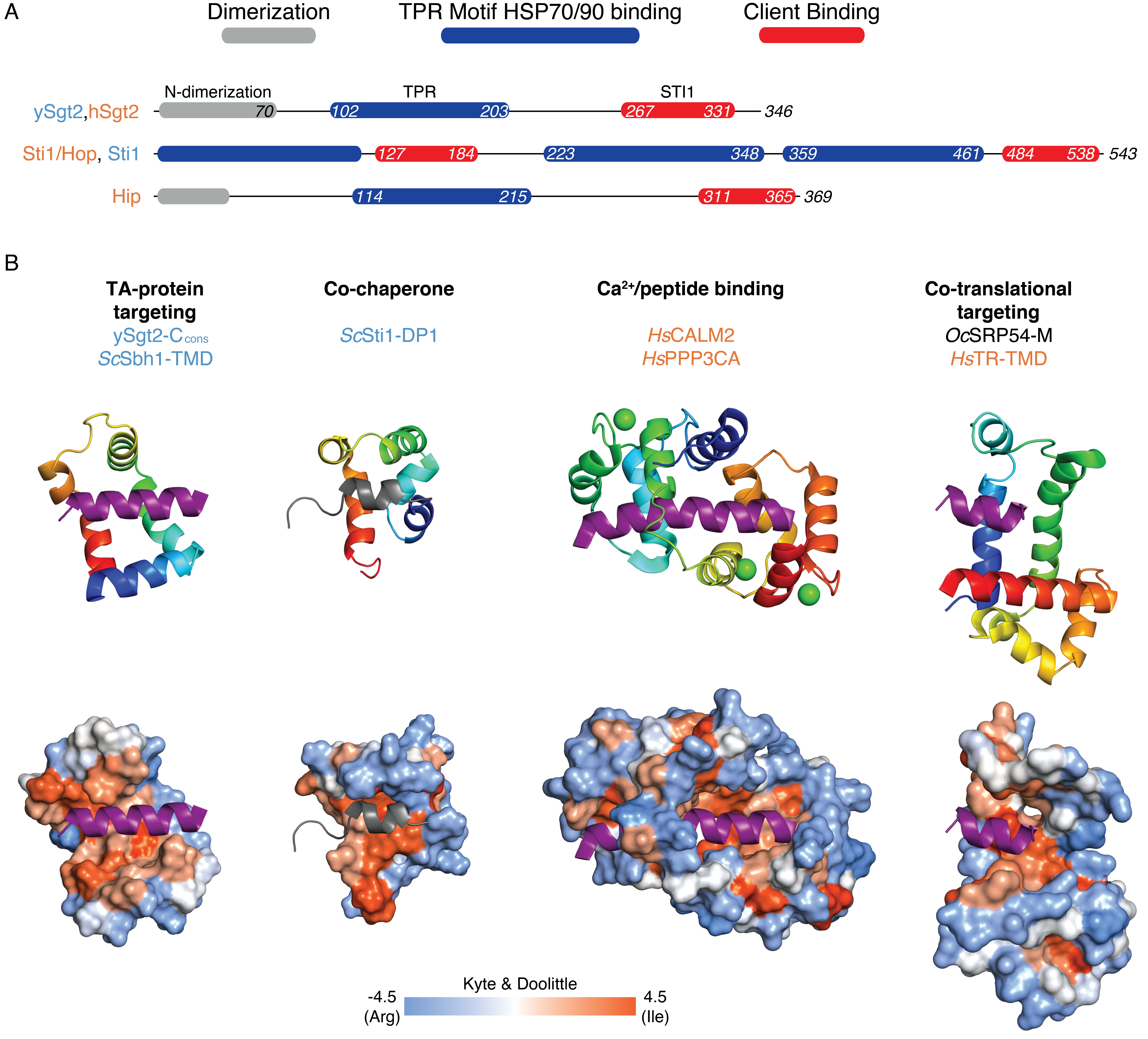
Computational modeling reveals that a conserved region, sufficient for client binding, forms a five alpha-helical hand which is reminiscent of other proteins involved in membrane protein targeting. Like Sgt2, the signal recognition particle (SRP) contains a methionine-rich domain that binds signal sequences and TMDs. While the helical order is inverted, again five amphipathic helices form a hydrophobic groove that cradles the client signal peptide (Voorhees and Hegde, 2015) (Figure 2.8B). Here once more, the domain has been observed to be flexible in the absence of client (Clemons et al., 1999; Keenan et al., 1998) and, in the resting state, occupied by a region that includes a helix which must be displaced (Voorhees and Hegde, 2015). Another helical-hand example recently shown to be involved in TA IMP targeting is calmodulin where a crystal structure reveals two helical hands coordinating to clasp a TMD at either end (Figure 2.8B) (Tidow and Nissen, 2013). Considering an average TMD of 18-20 amino acids (to span a ~40Å bilayer), each half of calmodulin interacts with about 10 amino acids. This is in close correspondence to the demonstrated minimal 11 amino acids for a TMD client to bind to the monomeric Sgt2-TPR-C. In the context of the full-length Sgt2, one can speculate that the Sgt2 dimer may utilize both C-domains to bind to a full TMD, similar to calmodulin. Cooperation of the two Sgt2 C-domains in client-binding could elicit conformational changes in the complex that could be recognized by downstream factors, such as additional interactions that increase the affinity to Get5/Ubl4A.
Intriguingly, Sgt2-TPR-C preferentially binds to artificial clients with clustered leucines. The hydrophobic groove presented in the computational model provides an attractive explanation for this preference. In order to bind to the hydrophobic groove, a client buries a portion of its TMD in the groove leaving the other face exposed. Clustering the most hydrophobic residues contributes to the hydrophobic effect driving binding efficiency and protecting them from the aqueous environment. Indeed, when focusing on Sgt2’s role in TA IMP targeting, GET pathway substrates have been suggested to be more hydrophobic TMDs than EMC substrates (Guna et al., 2018). Of these, for the most hydrophobic substrates, like Bos1, residues on both sides of the TMD could be protected by a pair of C-domains. Alternatively, the unstructured N-terminal loop through H0 could act as a lid surrounding the circumference of the client’s TMD. Unstructured regions participating in substrate binding as well as capping a hydrophobic groove have both suggested in the context of other domains, e.g., with Get3 (Guna and Hegde, 2018). The role for this clustering of hydrophobic residues in client recognition and targeting merits further investigation.
What is the benefit of the flexible helical-hand structure for hydrophobic helix binding? While it remains an open question, it is notable that evolution has settled on similar simple solutions to the complex problem of specific but temporary binding of hydrophobic helices. For all of the domains mentioned above, the flexible helical-hands provide an extensive hydrophobic surface to capture a client-helix—driven by the hydrophobic effect. Typically, such extensive interfaces are between pairs of pre-ordered surfaces resulting in high affinities and slow off rates. These helical hands are required to only engage temporarily, therefore the flexibility offsets the favorable free energy of binding by charging an additional entropic cost for ordering a flexible structure in the client-bound complex. The benefit for clients is a favorable transfer to downstream components in the GET pathway as seen for ySgt2 (Cho and Shan, 2018) and hSgt2 (Shao et al., 2017). The demonstration that TA client transfer from hSgt2 to Get3 is twice as fast as disassociation from hSgt2 into solution, perhaps interaction with Get3 leads to conformational changes that further favor release (Shao et al., 2017).
While hSgt2 and ySgt2 share many properties, there are a number of differences between the two homologs that may explain the different biochemical behavior. For the Ccons-domains, hSgt2 appears to be more ordered in the absence of client as the peaks in its NMR spectra are broader (Supplementary Figure 2.1B,C). Comparing the domains at the sequence level, while the high glutamine content in the C-domain is conserved it is higher in hSgt2 (8.8% versus 15.2%). The additional glutamines are concentrated in the predicted longer H4 helix (Figure 2.1A). The linker to the TPR domain is shorter compared to ySgt2 while the loop between H3 and H4 is longer. Do these differences reflect different roles? As noted, in every case the threshold for hydrophobicity of client-binding is lower for hSgt2 than ySgt2 (Figure 2.2E, Figure 2.6, Figure 2.7) implying that the mammalian protein is more permissive in client binding. The two C-domains have similar hydrophobicity, so this difference in binding might be due to a lower entropic cost paid by having the hSgt2 C-domain more ordered in the absence of client or the lack of an unstructured N-terminal loop.
The targeting of TA clients presents an intriguing and enigmatic problem for understanding the biogenesis of IMPs. How subtle differences in each client modulates the interplay of hand-offs that direct these proteins to the correct membrane remains to be understood. In this study, we focus on a central player, Sgt2 and its client-binding domain. Through biochemistry and computational analysis, we provide a structural model that adds more clarity to client binding both within and outside of the GET pathway.
2.5 Material and Methods
2.5.1 Plasmid constructs
MBP-Sbh1 full length, ySgt295-346 (ySgt2-TPR-C), ySgt2222-346 (ySgt2-C), ySgt2260-327 (ySgt2-Ccons), ySgt2266-327 (ySgt2-\(\Delta\)H0), hSgt287-313 (hSgt2-TPR-C), hSgt2213-313 (hSgt2-C), hSgt2219-300 (hSgt2-Ccons), and hSgt2228-300 (hSgt2-\(\Delta\)H0) were prepared as previously described (Chartron et al., 2011; Suloway et al., 2012). Genes of ySgt2 or hSgt2 variants were amplified from constructed plasmids and then ligated into an pET33b-derived vector with a 17 residue N-terminal hexa-histidine tag and a tobacco etch virus (TEV) protease site. Single or multiple mutations on Sgt2 were constructed by site-direct mutagenesis. Artificial clients were constructed in a pACYC-Duet plasmid with a N-terminal cMyc tag, BRIL fusion protein (Chu et al., 2002), GSS linker, and a hydrophobic C-terminal tail consisting of leucines and alanines and ending with a tryptophan.
2.5.2 Protein expression and purification
All proteins were expressed in Escherichia coli NiCo21 (DE3) cells (New England BioLabs). To co-express multiple proteins, constructed plasmids were co-transformed as described (Suloway et al., 2012). Protein expression was induced by 0.3 mM IPTG at OD600 ~ 0.7 and harvested after 3 hours at 37\(\degree\)C. For structural analysis, cells were lysed through an M-110L Microfludizer Processor (Microfluidics) in lysis buffer (50 mM Tris, 300 mM NaCl, 25 mM imidazole supplemented with benzamidine, phenylmethylsulfonyl fluoride (PMSF), and 10 mM \(\beta\)-mercaptoethanol (BME), pH 7.5). For capture assays, cells were lysed by freeze-thawing 3 times with 0.1 mg/mL lysozyme. To generate endogenous proteolytic products of ySgt2-TPR-C for MS analysis, PMSF and benzamidine were excluded from the lysis buffer. His-tagged Sgt2 and his-tagged Sgt2/TA complexes were separated from the lysate by batch incubation with Ni-NTA resin at 4\(\degree\)C for 1hr. The resin was washed with 20 mM Tris, 150 mM NaCl, 25 mM imidazole, 10 mM \(\beta\)ME, pH 7.5. The complexes of interest were eluted in 20 mM Tris, 150 mM NaCl, 300 mM imidazole, 10 mM \(\beta\)ME, pH 7.5.
For structural analysis, the affinity tag was removed from complexes collected after the nickel elution by an overnight TEV digestion against lysis buffer followed by size-exclusion chromatography using a HiLoad 16/60 Superdex 75 prep grade column (GE Healthcare).
Measurement of Sgt2 protein concentration was carried out using the bicinchoninic acid (BCA) assay with bovine serum albumin (BSA) as standard (Pierce Chemical Co.). Samples for NMR and CD analyses were concentrated to 10-15 mg/mL for storage at -80\(\degree\)C before experiments.
For the in vitro transfer assay, plasmids encoding for the full-length ySgt2 cysteine mutants were transformed into BL21 Star cells (Invitrogen). Cells were grown in 2x yeast-tryptone (2xYT) media and induced with 0.1mM IPTG at an OD600 of 0.6 then harvested after 3 hours at 30\(\degree\)C by centrifugation. Cells were lysed in 50mM Tris pH 8.0, 500mM NaCl, 10% glycerol, and 1x BugBuster (Millipore Sigma), supplemented with protease inhibitors (4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (Roche), benzamidine, and BME). Full-length his-tagged ySgt2 and cysteine mutants were separated from the lysate by batch incubation with Ni-NTA resin (Qiagen) at 4\(\degree\)C for 1 hour. The resin was washed with 50mM Tris pH 8.0, 500mM NaCl, 10% glycerol, and 25mM imidazole and then the protein was eluted in 50mM Tris pH 8.0, 500mM NaCl, 10% glycerol, and 300mM imidazole. For storage, protein was dialyzed in 25mM K-HEPES pH 7.5, 150mM KOAc, and 20% glycerol at 4\(\degree\)C and then flash frozen in liquid nitrogen. Purified Bos1 with p-benzoyl-l-phenylalanine (BPA) labeled at residue 230 (Bos1BPA) and yeast Ssa1 were gifts from the lab of Shu-ou Shan (Caltech).
2.5.3 NMR Spectroscopy
15N-labeled proteins were generated from cells grown in auto-induction minimal media as described (Studier, 2005) and purified in 20 mM phosphate buffer, pH 6.0 (for ySgt2-C, 10mM Tris, 100mM NaCl, pH 7.5). The NMR measurements of 15N-labeled Sgt2-C proteins (~0.3-0.5 mM) were collected using a Varian INOVA 600 MHz spectrometer at either 25\(\degree\)C (ySgt2-C) or 35\(\degree\)C (hSgt2-C) with a triple resonance probe and processed with TopSpin™ 3.2 (Bruker Co.).
2.5.4 CD Spectroscopy
The CD spectra were recorded at 24\(\degree\)C with an Aviv 202 spectropolarimeter using a 1 mm path length cuvette with 10 \(\mu\)M protein in 20 mM phosphate buffer, pH 7.0. The CD spectrum of each sample was recorded as the average over three scans from 190/195 to 250 nm in 1 nm steps. Each spectrum was then decomposed into its most probable secondary structure elements using BeStSel (Micsonai et al., 2015).
2.5.5 Glu-C digestion of the double cysteine mutants on ySgt2-C
Complexes of the co-expressed wild type or double cysteine mutated His-ySgt2-TPR-C and the artificial client, 11[L8], with either a cMyc or MBP tag were purified as the other His-Sgt2 complexes described above or initially purified via amylose affinity chromatography before nickel chromatography explained earlier. The protein complexes were mixed with 0.2 mM CuSO4 and 0.4 mM 1,10-phenanthroline at 24\(\degree\)C for 20 min followed by 50 mM N-ethyl maleimide for 15 min. Sequencing-grade Glu-C protease (Sigma) was mixed with the protein samples at an approximate ratio of 1:30 and the digestion was conducted at 37\(\degree\)C for 22 hours. Digested samples were mixed with either non-reducing or reducing SDS-sample buffer, resolved via SDS-PAGE using Mini-Protean® Tris-Tricine Precast Gels (10-20%, Bio-Rad), and visualized using Coomassie Blue staining.
2.5.6 In vitro transfer assay of Bos1 from Ssa1 to ySgt2
The in vitro transfer assays were performed as in Chio et al. 2019 and Shao et al. 2017 (Chio et al., 2019; Shao et al., 2017). Specifically, 39µM Bos1BPA (50mM HEPES, 300mM NaCl, 0.05% LDAO, 20% glycerol) was diluted to a final concentration of 0.1µM and added to 4µM Ssa1 supplemented with 2mM ATP (25mM HEPES pH7.5, 150mM KOAc). After one minute, 0.3µM of full-length ySgt2 or mutant was added to the reaction. Samples were flash frozen after one minute and placed under a 365nm UV lamp for 2 hours on dry ice to allow for BPA crosslinking.
2.5.7 Protein immunoblotting and detection
For western blots, protein samples were resolved via SDS-PAGE and then transferred onto nitrocellulose membranes by the Trans-Blot® Turbo™ Transfer System (Bio-Rad). Membranes were blocked in 5% non-fat dry milk and hybridized with antibodies in TBST buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.1% Tween 20) for 1 hour of each step at 24\(\degree\)C. The primary antibodies were used at the following dilutions: 1:1000 anti-penta-His mouse monoclonal (Qiagen), 1:5000 anti-cMyc mouse monoclonal (Sigma), and a 1:3000 anti-Strep II rabbit polyclonal (Abcam). A secondary antibody conjugated either to alkaline phosphatase (Rockland, 1:8000) or a 800nm fluorophore was employed, and the blotting signals were chemically visualized with either the nitro-blue tetrazolium/5-bromo-4-chloro-3’-indolyphosphate (NBT/BCIP) chromogenic assay (Sigma) or infrared scanner. All blots were photographed and quantified by image densitometry using ImageJ (Schneider et al., 2012) or ImageStudioLite (LI-COR Biosciences).
2.5.8 Quantification of Sgt2-TA complex formation
The densitometric analysis of MBP-Sbh1 capture by His-Sgt2-TPR-C quantified the intensity of the corresponding protein bands on a Coomassie Blue G-250 stained gel. The quantified signal ratios of MBP-Sbh1/His-Sgt2-TPR-C are normalized to the ratio obtained from the wild-type (WT). Expression level of MBP-Sbh1 was confirmed by immunoblotting the MBP signal in cell lysate. Average ratios and standard deviations were obtained from 3-4 independent experiments.
In artificial client experiments, both his-tagged Sgt2-TPR-C and cMyc-tagged artificial clients were quantified via immunoblotting signals. The complex efficiency of Sgt2-TPR-C with various clients was obtained by
\(\text{E}\text{complex}\text{ }\text{= }\frac{\text{E}_{\text{TMD}}}{\text{T}_{\text{T}\text{MD}}}\text{×}\frac{\text{1}}{\text{E}_{\text{capture}}}\) (1)
where ETMD is the signal intensity of an eluted client representing the amount of client co-purified with Sgt2-TPR-C and TTMD is the signal intensity of a client in total lysate which corresponds to the expression yield of that client. Identical volumes of elution and total lysate from different clients experiments were analyzed and quantified. In order to correct for possible variation the amount of Sgt2-TPR-C available for complex formation, Ecapture represents the relative amount of Sgt2-TPR-C present in the elution (ESgt2) compared to a pure Sgt2-TPR-C standard (Epurified,Sgt2).
\(\text{E}\text{capture}\text{ }\text{= }\frac{\text{E}_{\text{Sgt2}}}{\text{E}_{\text{purified,Sgt2}}}\text{,}\) (2)
Each ETMD and TTMD value was obtained by blotting both simultaneously, i.e., adjacently on the same blotting paper. To facilitate comparison between clients, the Sgt2-TPR-C/TA client complex efficiency Ecomplex,TMD is normalized by Sgt2-TPR-C/Bos1 complex efficiency Ecomplex,Bos1.
\(\text{\% Complex = }\frac{\text{E}_{\text{complex,T}\text{MD}}}{\text{E}_{\text{complex,B}\text{os}\text{1}}}\text{ × 100}\) (3)
2.5.9 Sequence alignments
An alignment of Sgt2-C domains was carried out as follows: all sequences with an annotated N-terminal Sgt2/A dimerization domain (PF16546 (Finn et al., 2016)), at least one TPR hit (PF00515.27, PF13176.5, PF07719.16, PF13176.5, PF13181.5), and at least 50 residues following the TPR domain were considered family members. Putative C-domains were inferred as all residues following the TPR domain, filtered at 90% sequence identity using CD-HIT (Li and Godzik, 2006), and then aligned using MAFFT G-INS-I (Katoh and Standley, 2013). Other attempts with a smaller set (therefore more divergent) of sequences results in an ambiguity in the relative register of H0, H1, H2, and H3 when comparing Sgt2 with SGTA.
Alignments of Sti1 (DP1/DP2) and STI1 domains were created by pulling all unique domain structures with annotated STI1 domains from Uniprot. Where present, the human homolog was selected and then aligned with PROMALS3D (Pei et al., 2008). PROMALS3D provides a way of integrating a variety of costs into the alignment procedure, including 3D structure, secondary structure predictions, and known homologous positions.
All alignments were visualized using Jalview (Waterhouse et al., 2009).
2.5.10 Molecular modeling
Putative models for ySgt2-C were generated with I-TASSER, PCONS, Quark, Robetta (ab initio and transform-restrained modes), Phyre2, and RaptorX via their respective web servers (Bradley et al., 2005; Wallner and Elofsson, 2006; Xu and Zhang, 2012; Yang et al., 2020, 2015). The highest scoring model from Quark was then chosen to identify putative TA client binding sites by rigid-body docking of various transmembrane domains modelled as \(\alpha\)-helices (3D-HM (Reißer et al., 2014)) into the ySgt2-Ccons through the Zdock web server (Pierce et al., 2014). Pairwise distances were calculated between C\(\beta\) atoms (the closer C\(\alpha\) proton on glycine) using mdtraj (McGibbon et al., 2015). Based on our disulfide crosslinks, new models were predicted using Robetta in ab initio mode specifying C\(\beta\)-C\(\beta\) atom distance constraints bounded between 0 and 9 Å.
For hSgt2, using the same set of structure prediction servers above, we were only able to produce a clear structural model using the Robetta transform-restrained mode. We were also unable to generate a reliable model by directly using the ySgt2-C model as a template (Webb and Sali, 2016). To crosslink distance data from ySgt2 as restraints for hSgt2, pair positions were transferred from one protein to the other via an alignment of Sgt2-C domains (excerpt in Figure 2.1A) and ran Robetta ab initio. Also, we grafted the N-terminal loop of ySgt2-C on hSgt2-C with the same set of restraints.
Images were rendered using PyMOL 2.3.
2.6 Supplementary Figures